Jan. 29, 2010 Press Release Biology Computing / Math
Simulating the cell, one molecule at a time
A novel technique for ultrahigh-precision simulation of cellular dynamics has enabled researchers to uncover a mechanism underlying a ubiquitous response process in eukaryotic cells. The findings, to appear in the upcoming issue of the Proceedings of the National Academy of Sciences (PNAS), mark a first step toward the full-scale molecular-level simulation of biological cells.
Signaling processes in cells depend on a complex web of interactions whose macro-level behavior arises from micro-level dynamics of molecules. In mitogen-activated protein kinase (MAPK) cascades, a key pathway for signaling, such dynamics have been shown to generate rich phenomena such as ultrasensitivity and bistability that are critical to biological function. Conventional techniques for modeling MAPK, however, ignore such micro-scale dynamics to reduce computational time.
With their latest work, researchers at RIKEN and the FOM Institute for Atomic and Molecular Physics (AMOLF), Holland, have sidestepped this problem. The group applied a technique they developed, enhanced Green Function Reaction Dynamics (eGFRD), to simulate particle-level dynamics of the MAPK system, dramatically reducing computation time. Their results reveal that micro-level details of the interaction between enzyme and substrate molecules, overlooked in earlier models, can have a dramatic impact on the nature of response.
While challenging conventional theory, the findings also prelude an era of cellular simulations on a level of molecular detail never before possible. By exploiting the power of RIKEN's Next- Generation Supercomputer, currently under construction in Kobe, the new technique opens the door to the full-scale simulation of living cells, with key applications to cancer and stem cell research.
Contact
Koichi Takahashi
Biochemical Simulation Research Team, Computational Systems Biology Research Group
Advanced Computational Sciences Department
RIKEN Advanced Science Institute
Tel: +81-(0)45-503-9430 / Fax: +81-(0)45-503-9429
Jens Wilkinson
RIKEN Global Relations and Research Coordination Office
Tel: +81-(0)48-462-1225 / Fax: +81-(0)48-463-3687
Email: pr@riken.jp
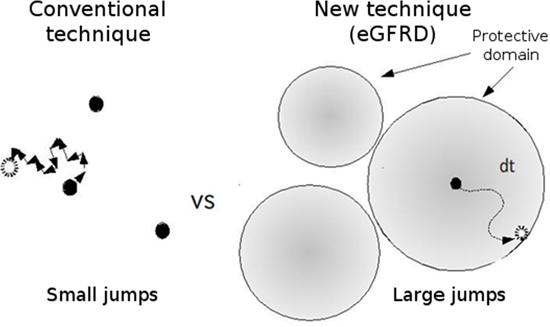
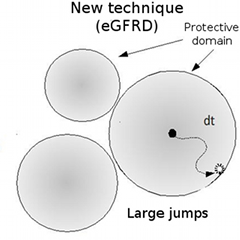
Figure 1: Comparison of conventional and eGFRD simulation techniques
With the eGFRD technique, the region within which particles can move freely is defined, enabling the simulation to proceed across a large distance in only a single jump.
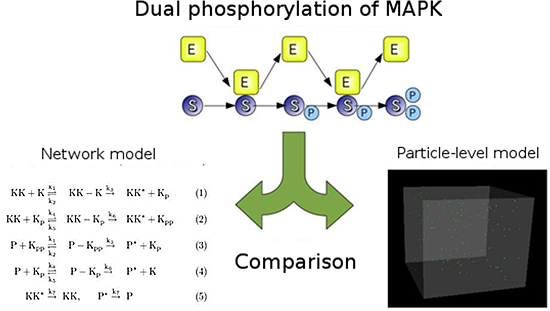
Figure 2: MAPK simulation
Top: MAPK molecules (S), MAPKK molecules (E) and dual phosphorylation (P).
Bottom: Comparison of dual phosphorylation of MAPK as simulated by a network model which does not take into consideration the spatial motion of particles (left) and by a fine-grained spatial particle-based model (right).
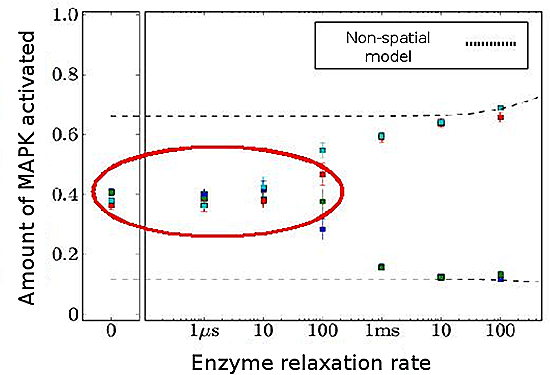
Figure 3: Results of MAPK simulation
As relaxation rate of enzymes increases, bistability response disappears in some cases (red circle).