Jun. 14, 2010 Press Release Biology
RIKEN and SISSA develop new biological analysis methods
Researchers in Japan and Italy have developed new methods for biological analysis with dramatically higher sensitivity and detail than earlier techniques, expanding our understanding of RNA and its function in the cell. Described in the June 13th issue of Nature Methods, the methods open the door to the study of diseases well beyond the reach of current technologies.
Transcription, the process of creating an RNA copy from a DNA sequence, is the first step in the process of gene expression, resulting in the creation of proteins or formation of non-coding RNA. In recent years, the study of mammalian RNA transcripts has revealed a richness of complexity in transcript structure and function, while enabling researchers to characterize cancers and other diseases in more detail than ever before.
The two new methods developed, nanoCAGE and CAGEscan, greatly enhance this detail by extending the capabilities of Cap Analysis of Gene Expression (CAGE), a high-throughput method for profiling RNA transcripts. Developed by researchers with Dr. Piero Carninci at the RIKEN Omics Science Center and Dr. Stefano Gustincich at SISSA, the new methods link transcript start sites (TSSs) in genes, where RNA transcription begins, with the set of RNA molecules transcribed.
With a level of sensitivity one thousand times higher than CAGE, nanoCAGE presents powerful new possibilities for the analysis of small-sized samples. Using nanoCAGE, as few as 1000 cells is enough to detect all RNA transcripts in a cell and map the genomic regions that cause a gene to be expressed, providing a detailed picture of how gene expression is regulated. nanoCAGE thus overcomes the difficulty of obtaining large quantity of cells for studying and future diagnosing diseases such as cancer.
CAGEscan complements nanoCAGE by pinpointing the gene's product - the RNA molecule - from the TSS where transcription begins. CAGEscan presents an instrumental tool for exploring the "RNA continent", thousands of RNAs that do not code for proteins but function to modulate the cell's genetic program. While presenting promising targets for new therapeutic approaches, these RNA have remained obscure in the past due to the limitations of existing technologies.
The two new methods greatly enhance the ability of researchers to work with tiny samples and improve the efficiency of RNA identification. Simple to perform and requiring no specialized equipment, nanoCAGE will be the technology of choice for molecular biologists, while CAGEscan will accelerate research in domains where our current knowledge of active genes is limited. Together, the two methods set the stage for new applications in drug screening, biopsy analysis and whole-transcriptome association studies.
Reference
- Charles Plessy, Nicolas Bertin, Hazuki Takahashi, Roberto Simone, Md Salimullah, Timo Lassmann, Morana Vitezic, Jessica Severin, Signe Olivarius, Dejan Lazarevic, Nadine Hornig, Valerio Orlando, Ian Bell, Hui Gao, Jacqueline Dumais, Philipp Kapranov, Huaien Wang, Carrie A Davis, Thomas R Gingeras, Jun Kawai, Carsten O Daub, Yoshihide Hayashizaki, Stefano Gustincich & Piero Carninci. "Linking promoters to functional transcripts in small samples with nanoCAGE and CAGEscan". Nature Methods 7, 528 - 534 (2010). doi: 10.1038/nmeth.1470
Contact
Dr. Piero Carninci
Functional Genomics Technology Team
RIKEN Omics Science Center
Tel: +81-(0)45-503-9222 / Fax: +81-(0)45-503-9216
Dr. Stefano Gustincich
Group Head of Neurobiology Sector SISSA
Jens Wilkinson
RIKEN Global Relations and Research Coordination Office
Tel: +81-(0)48-462-1225 / Fax: +81-(0)48-463-3687
Email: pr@riken.jp
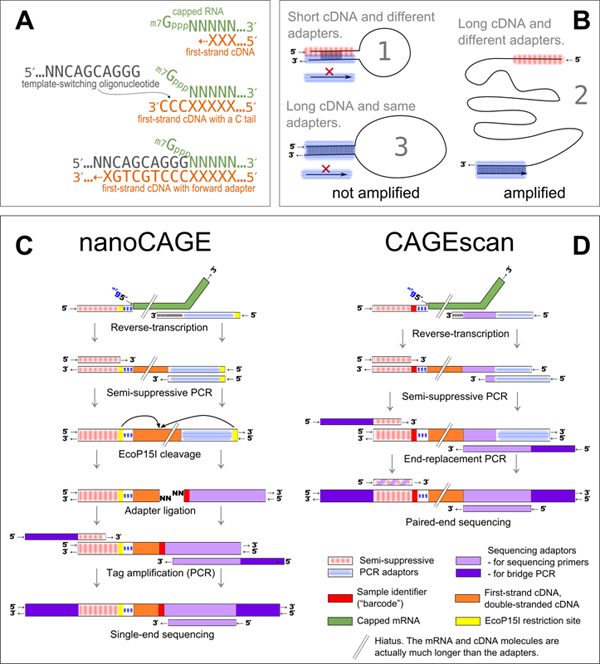
Experimental outline of the nanoCAGE and CAGEscan protocols
A: nanoCAGE captures the 5' ends of molecules by template switching. When polymerizing the cDNA of a capped mRNA, the reverse transcriptase adds extra cytosines that are complementary to the cap. Each 5' full-length cDNAs is extended upon hybridization of the riboguanosine-tailed template-switching oligonucleotides to these extra cytosines.
B: In the semisuppressive PCR, the short templates fold intramolecularly and prevent the binding of primers which precludes amplification; longer molecules are less likely to fold and are thus amplified. Templates derived from reaction artifacts form stable homoduplexes, also precluding amplification.
C: Preparation of nanoCAGE tags. After template-switching, semisuppressive PCR and EcoP151 cleavage, 25-bp tags are ligated to barcode-containing oligonucleotide adapters. After PCR amplification, the nanoCAGE tags are sequenced by synthesis.
D: Preparation of 5'-full-length cDNA libraries for paired-end sequencing with the CAGEscan protocol. Capped mRNAs capture is similar to that described in a. The ends of the amplified cDNA constructs are replaced in a PCR with adapters for sequencing in the Illumina Genome Analyzer, which produces paired-end reads from single cDNAs. Line breaks indicate that mRNA and cDNA are much longer than adaptors.