Dec. 9, 2010 Press Release Biology
New study uncovers how cellular stress causes brain damage
New findings by researchers at RIKEN, Japan's flagship research institution, have linked a specific type of cellular stress to neuronal cell death leading to brain damage. Published in the journal Neuron, the findings overturn existing assumptions on the role of a key neuronal protein in cellular stress response, opening up new avenues for research on a range of neurodegenerative diseases.
As an organelle responsible for the production, processing and transport of a wide variety of cellular materials, the endoplasmic reticulum (ER) plays a central role in maintaining protein quality in the cell. Pathological conditions that affect protein folding or calcium signaling can interfere with this role, causing stress to the ER which, in severe cases, can trigger cell death (apoptosis). In the brain, such apoptosis has been associated with neurodegenerative diseases such as Alzheimer's disease and Huntington's disease (HD), yet the mechanisms involved remain poorly understood.
To clarify these mechanisms, the researchers investigated the relationship between ER stress and a neuronal protein called inositol 1,4,5-trisphosphate receptor 1 (IP3R1), one of three IP3R receptors that modulate intracellular calcium signaling. Using calcium imaging techniques, the team identified a sharp decline in IP3R1 activity in cells treated with ER stress inducers. It was further revealed that the ER stress-dependent dysfunction of IP3R1 induced neuronal cell death and brain damage, situating IP3R1 as a crucial link between ER stress and neuron cell death.
Underlying this link, the researchers identified a mechanism through which GRP78, a molecular chaperone, binds to a region of IP3R1 called L3V to positively regulate tetrameric assembly of IP3R1. ER stress, they show, impairs this assembly mechanism and subsequently inhibits IP3R1 activation, a process also observed in the brain of model mice with HD.
As the first research to highlight the significant role of IP3R1 in protecting the brain from ER stress, the Neuron study marks a major step toward clarifying the mechanisms underlying stress-induced brain damage, promising advancements in the treatment of neurodegenerative diseases.
Reference
- Takayasu Higo, Kozo Hamada, Chihiro Hisatsune, Nobuyuki Nukina, Tsutomu Hashikawa, Mitsuharu Hattori, Takeshi Nakamura and Katsuhiko Mikoshiba. Mechanism of ER Stress-Induced Brain Damage by IP3 Receptor. Neuron 68(5): 865-878. DOI: 10.1016/j.neuron.2010.11.010
Contact
Katsuhiko Mikoshiba
Laboratory for Developmental Neurobiology
RIKEN Brain Science Institute
Jens Wilkinson
RIKEN Global Relations and Research Coordination Office
Tel: +81-(0)48-462-1225 / Fax: +81-(0)48-463-3687
Email: pr@riken.jp
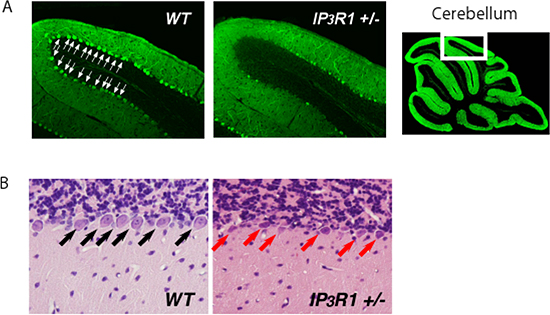
Figure 1: Loss of IP3R1 enhances ER stress-induced neuronal cell death.
Representative anti-calbindin-stained (A) and hematoxylin and eosin-stained (B) cerebellum sections from WT and IP3R1 +/- mice sacrificed 2 days after injection of an ER stressor, tunicamycin. Note marked decrease of calbindin signals (A) and increase of shrunken neurons (B) in the cerebellum lobule VI of IP3R1 +/-.
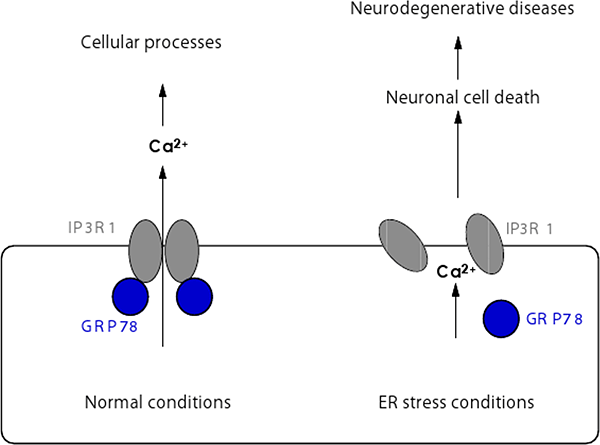
Figure 2: Regulation of tetrameric assembly of IP3R1 under normal and ER stress conditions.
GRP78 positively regulates tetrameric assembly of IP3R1 in normal conditions, enabling IP3R1 to function as a calcium release channel regulating various cellular processes. In contrast, under ER stress conditions the functional interaction between IP3R1 and GRP78 is impaired which attenuates intracellular calcium signals to cause cell death and brain damage.