Dec. 9, 2016 Research Highlight Biology
Mouse study throws doubt on hallmarks of Alzheimer’s disease
Many previous studies on the mouse modeling of Alzheimer’s disease may have been confounded by inadequate genetic engineering used in experiments
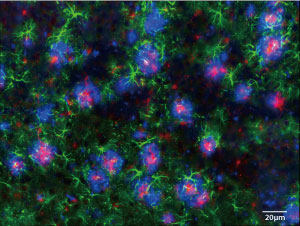 Figure 1: A fluorescence microscopy image of the brain of the first knock-in mice produced for studying Alzheimer’s disease. The blue areas indicate the accumulation of amyloid beta peptide in the brain. © 2016 Takaomi Saido, RIKEN Brain Science Institute
Figure 1: A fluorescence microscopy image of the brain of the first knock-in mice produced for studying Alzheimer’s disease. The blue areas indicate the accumulation of amyloid beta peptide in the brain. © 2016 Takaomi Saido, RIKEN Brain Science Institute
The findings of more than 3,000 studies on Alzheimer’s disease have been called into question by a new RIKEN study. It suggests that two characteristics previously believed to be important in defining the disease may be side effects of the creation of genetically engineered mice that mimic the condition1.
Humans are the only animals known to succumb to Alzheimer’s disease. To gain insights into the disease, researchers engineer mice that carry human mutations associated with Alzheimer’s. But so far, no one has produced mice that exhibit the disease.
The closest researchers have come is to produce mice with highly elevated levels of mutant amyloid precursor protein (APP). These mice exhibit pathological hallmarks of Alzheimer’s, including the accumulation of amyloid beta peptide in the brain (Fig. 1), which is widely believed to cause the disease. This accumulation is usually accompanied by increased levels of a protein called p25 and a reduction in a sodium-ion channel known as Nav1.1, both of which are thought to play important roles in the disease’s development.
Typically, mutant mice created for Alzheimer studies carry multiple copies of the human gene that carries the instructions for making mutant APP. Takaomi Saido and colleagues at the RIKEN Brain Science Institute have succeeded in creating the first ‘knock-in’ mice where only one copy of the mutated APP gene is inserted at a targeted location in the genome.
 Figure 2: Takaomi Saido and his team have found that the results obtained from mice studies of Alzheimer's disease may have been affected by the kind of experimental mice used. © 2016 RIKEN
Figure 2: Takaomi Saido and his team have found that the results obtained from mice studies of Alzheimer's disease may have been affected by the kind of experimental mice used. © 2016 RIKEN
The team compared these knock-in mice, which synthesized physiologically normal amounts of human APP, with mice that are typically used in Alzheimer’s research. All the animals accumulated amyloid beta peptide, but the knock-in mice with normal levels of mutated human APP did not exhibit overproduction of p25 or a reduction in Nav1.1.
This suggests that some of the characteristics of mouse models of Alzheimer’s could be experimental quirks, or artifacts. To confirm this, the researchers created another strain of mutant mice that produced normal levels of mutated human APP but lacked the gene encoding calpastatin, an enzyme that normally inhibits the production of p25. With calpastatin erased, researchers expected p25 to increase, but these animals did not overproduce p25 either. The researchers concluded that the altered p25 and Nav1.1 levels seen in mice that overproduce mutant human APP arise chiefly because of artificial overproduction of mutant APP.
“We have about 200 collaborators around the world that have started using our mice, so they may find other types of artifacts,” says Saido. “But we are more interested in facts than artifacts, and so we are mainly working on the mechanisms of neurodegeneration.”
Related contents
- New mouse model could revolutionize research in Alzheimer's disease
- New mouse models raise the bar for Alzheimer’s research
- Alzheimer's plaques reduced by targeting sugar attachment to the BACE1 enzyme
References
- 1. Saito, T., Matsuba, Y., Yamazaki, N., Hashimoto, S. & Saido, T. C. Calpain activation in Alzheimer’s model mice is an artifact of APP and presenilin overexpression. Journal of Neuroscience 36, 9933–9936 (2016). doi: 10.1523/jneurosci.1907-16.2016