2010年7月9日
シンシナティ大学
独立行政法人 理化学研究所
独立行政法人 科学技術振興機構
IP3レセプターは、心不全治療の新しいターゲット
-IP3レセプターを介するカルシウムイオン流出が心肥大の原因に-
ポイント
- 生きたマウスの心臓で、II型IP3レセプター活性化が心肥大を引き起こすことを確認
- IP3レセプター活性化の抑制が、アンジオテンシンIIなどによる心肥大を防ぐ
- 心肥大を伴う心不全の新規治療薬開発へ第一歩
要旨
米国シンシナティ大学(グレゴリー・ウィリアムズ学長)と独立行政法人理化学研究所(野依良治理事長)は、生きたマウスの心臓(生体心)を用いて、慢性心不全の主原因とされる心肥大の形成に、細胞内カルシウムイオンチャネルであるイノシトール三リン酸受容体(IP3レセプター)※1がかかわっていることを明らかにしました。シンシナティ大学の中山博之研究員(現、大阪大学大学院薬学研究科准教授)、ジェフリー・モルケンティン(Jeffery Molkentin)教授と理研脳科学総合研究センター(利根川進センター長)発生神経生物研究チームの御子柴克彦チームリーダー(独立行政法人科学技術振興機構戦略的創造研究推進事業 発展研究(SORST)「カルシウム振動プロジェクト」研究総括)らによる共同研究の成果です。
慢性心不全は欧米の先進国における死亡原因第一位の疾患で、その病態の進行には心肥大がかかわっていると考えられています。細胞内のカルシウムイオンは、心収縮制御に重要な役割を果たしていますが、慢性心不全では、収縮期の細胞内カルシウムイオン濃度は低下しています。一方で、カルシニューリン※2などのカルシウムイオン依存性のシグナル分子が活性化し、心肥大形成を引き起こすことが知られています。しかし、心筋細胞ではカルシウムイオン濃度の変動が大きく、カルシウムイオンが心肥大形成を引き起こすメカニズムは長い間不明でした。
研究グループは、心肥大形成のメカニズムとして、心収縮を制御するカルシウムイオンとは別に、IP3レセプターを介するカルシウムイオンの流出が起こると考えました。そこで、心臓で優位に発現しているとされるII型IP3レセプターの過剰発現マウスを作製し、IP3を生成する刺激を加えたところ、IP3レセプターを介してカルシウムイオンが流出し、心肥大反応が増大することを見いだしました。また、心筋で特異的にIP3レセプターの活性を抑制するマウスを作製し、心肥大反応が減弱することを確認しました。
心肥大形成にIP3レセプターを介したカルシウム流出が重要な役割を果たすことが判明したことで、新しい心不全治療薬の開発に大きく貢献すると期待されます。
本研究成果は、米国の科学雑誌『Circulation Research』(9月3日号)に掲載されるに先立ち、オンライン版(7月8日付け:日本時間7月9日)に掲載されます。
背景
慢性心不全は、欧米の先進国において死亡原因の第一位を占める治療の困難な疾患で、心臓の収縮拡張障害を主たる臨床的特徴としています。慢性心不全では、心肥大や心筋組織の線維化が観察され、これらの病理学的変化が病態の進行に深くかかわっていると考えられています。カルシウムイオンは、心筋細胞の興奮収縮連関※3における重要なセカンドメッセンジャーとして、細胞内濃度変化により心拍ごとの収縮と弛緩を制御しています。その一方で、カルシニューリンやカルモデュリン依存性キナーゼなどのカルシウム依存性シグナル分子を活性化することで、心肥大や細胞死のような病理学的変化でも中心的な役割を果たしています。しかし、心筋細胞のように常に細胞内カルシウムイオン濃度が変化する細胞では、これらのカルシウム依存性シグナルの活性化により心肥大を引き起こすメカニズムは不明でした(図1)。研究グループは、特定の分子からのカルシウムイオン流出による局所的なカルシウムイオン濃度の上昇が、心肥大を引き起こすと考え、イノシトール1,4,5-三リン酸受容体(IP3レセプター)に注目しました。
アンジオテンシンII受容体やβアドレナリン受容体などのGタンパク質共役受容体※4は、刺激を受けると心肥大を引き起こすため、慢性心不全の病態形成において重要な役割を果たしていることが知られています。心臓では、これらの受容体を刺激するとIP3を生成します。このIP3によってIP3レセプターが活性化され、細胞内のカルシウム貯蔵庫である小胞体からカルシウムイオンを放出します。IP3レセプターには3つのタイプがあり、いずれも心臓で発現していますが、中でもII型IP3レセプターの発現が優位であると考えられています。
そこで、Gタンパク質共役受容体刺激による心肥大形成で、IP3レセプターがどのような役割を果たしているか、生きたマウスの心臓(生体心)で解明することに挑みました。
研究手法と成果
(1)心筋特異的II型IP3レセプター過剰発現マウスの作製と解析
心筋特異的にII型IP3レセプターを過剰発現するマウスを作製し、表現型を解析したところ、12週齢で心肥大を示しました。このマウスから単離した成体心筋細胞を用いて、IP3レセプターを介するカルシウムイオンの流出を調べました。Gタンパク質共役受容体の1つであるエンドセリン受容体を刺激してIP3を生成すると、カルシウムトランジエント※5の増大を観察しました。この結果から、過剰発現したII型IP3レセプターがIP3感受性カルシウムチャネルとして機能していることが分かりました。
心筋細胞においてIP3を生成するアンジオテンシンII受容体持続刺激やβアドレナリン受容体持続刺激によっても、II型IP3レセプター過剰発現マウスは過大な心肥大反応を示しました。これらの結果は、II型IP3レセプターを介するカルシウムイオンの流出が、生体における心肥大を引き起こすことを示しています。
(2)心筋特異的IP3スポンジ※6過剰発現マウスの作製と解析
心筋特異的にIP3スポンジを過剰発現し、心筋におけるIP3レセプターを介するカルシウムイオンの流出を抑制するマウスを作製しました。このマウスでエンドセリン受容体刺激によりIP3を生成すると、II型IP3レセプターからのカルシウムイオン流出を抑制しました。これは、エンドセリン刺激で生成したIP3がIP3スポンジに捕捉され、II型IP3レセプターの活性化が抑制されていることを示しています。
IP3スポンジ過剰発現マウスは、アンジオテンシンII受容体持続刺激やβアドレナリン受容体持続刺激に対しても、肥大反応の減弱を示しました。これらの結果から、II型IP3レセプターの活性化を抑制すると、生体における心肥大が抑えられることを確認できました。
(3)生体心におけるIP3レセプターによる心肥大へのカルシニューリンの関与
転写因子NFAT(Nuclear factor of activated T-cells)※7は、カルシニューリンによって活性化され、心肥大を引き起こします。IP3レセプターを介したカルシウムイオンの流出による心肥大に、カルシニューリンが関与しているかどうかを、II型IP3レセプター過剰発現マウスで調べました。このマウスでNFATの転写活性を測定したところ、転写活性の上昇が認められ、カルシニューリンが活性化していることが分かりました。これは、II型IP3レセプターからのカルシウムイオンの流出がカルシニューリンを活性化し、心肥大を引き起こす可能性を示しています。
そこで、カルシニューリンノックアウトマウスを用いて、IP3レセプターを介したカルシウムイオンの流出による心肥大に、カルシニューリンが関与しているかどうかを調べました。カルシニューリンを欠損したII型IP3レセプター過剰発現マウスに、Gタンパク質共役受容体刺激として持続カテコラミン刺激を加えたところ、心肥大反応は起こりませんでした。この結果から、II型IP3レセプターを介するカルシウムイオン流出がカルシニューリンを活性化して、心肥大を引き起こすことが証明できました(図2)。
今後の期待
現在、慢性心不全の予後を改善するのに有効な治療薬として、β受容体遮断薬とアンジオテンシン変換酵素阻害薬が有効とされています。この2つの薬剤は、Gタンパク質共役受容体刺激を抑制することでその効果を発揮します。今回、Gタンパク質共役受容体刺激が引き起こす心肥大において、IP3レセプターが重要な役割を果たしていることが判明しました。心筋細胞で、これらの刺激と最も密接にかかわるIP3レセプターの機能を制御することで、心不全の予後を改善する新しい薬剤の開発が期待されます。
発表者
国立大学法人大阪大学大学院
薬学研究科臨床薬効解析学分野
准教授 中山 博之(なかやま ひろゆき)
Tel: 06-6879-8252 / Fax: 06-6879-8253
脳科学総合研究センター 疾患メカニズムコア 発生神経生物研究チーム
チームリーダー 御子柴 克彦(みこしば かつひこ)
Tel: 048-467-9745 / Fax: 048-467-9744
お問い合わせ先
脳科学研究推進部 企画課Tel: 048-467-9757 / Fax: 048-462-4914
(JSTの事業に関すること)
独立行政法人科学技術振興機構
イノベーション推進本部 研究プロジェクト推進部
小林 正(こばやし ただし)
Tel: 03-3512-3528 / Fax: 03-3222-2068
報道担当
独立行政法人理化学研究所 広報室 報道担当
Tel:048-467-9272 / Fax:048-462-4715
独立行政法人科学技術振興機構 広報ポータル部
Tel: 03-5214-8404 / Fax: 03-5214-8432
補足説明
- 1.イノシトール三リン酸受容体(IP3レセプター)
細胞内カルシウムイオンチャネル。細胞外情報物質(ホルモンや神経伝達物質)が細胞膜にある受容体に結合した結果、細胞膜の構成成分の1つであるホスファチジルイノシトール二リン酸が、ホスホリパーゼCによって加水分解されて細胞内にIP3を生じる。IP3は代表的な細胞内シグナル伝達分子で、Ca2+貯蔵庫である小胞体から細胞質へのCa2+の放出を促進する。 - 2.カルシニューリン
カルシウム/カルモデュリン依存的に活性化する脱リン酸化酵素。カルシウムイオン濃度の上昇によってカルモデュリンが構造変化を起こし、カルシニューリンに結合することで活性化する。心筋特異的過剰発現により心肥大と心不全を引き起こす。 - 3.興奮収縮連関
心筋、骨格筋、平滑筋などが筋収縮する際の、細胞膜の電気的変化(興奮)から筋収縮に至るまでの過程。 - 4.Gタンパク質共役受容体
Gタンパク質と相互作用することで情報伝達を行う受容体で、7回膜貫通型受容体とも呼ばれる。Gタンパク質はα、β、γの3種類のタンパク質による三量体構造をとる。受容体刺激によりGαが解離しシグナルを伝達する。 GαはGαs, Gαq, GαiおよびGα12の4つのファミリーに分類され、それぞれ特異的なセカンドメッセンジャーにシグナルを伝える。 - 5.カルシウムトランジエント
細胞内カルシウムイオンの濃度の一過性の上昇と低下により形成される波。カルシウムサイクリングと呼ばれる機構により形成される。 - 6.IP3スポンジ
生成されたIP3そのものを不活化する合成タンパク質。2002年に理研の御子柴克彦チームリーダーらが開発した。IP3スポンジはIP3レセプターと比較して約500倍のIP3に対する親和性を有し、IP3を結合することによりIP3レセプターの活性化を抑制する。 - 7.NFAT(Nuclear factor of activated T-cells)
転写因子。脱リン酸化により細胞質から核内に移行し、転写活性を示す。心筋細胞ではカルシニューリンにより脱リン酸化され核内に移行後、心肥大関連遺伝子の転写を促進する。
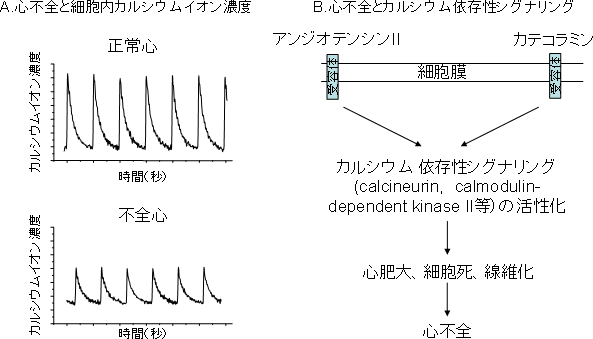
図1 心不全でのカルシウムイオン依存性シグナリングの活性化
(A) 1つ1つのスパイク状の波がカルシウムトランジエントを表す。心筋細胞は、興奮収縮連関により細胞膜の興奮を収縮力に変換する。カルシウムイオンは重要なセカンドメッセンジャーとして興奮収縮連関を制御している。心不全では、収縮期のカルシウムイオン濃度の低下に伴い、心筋細胞の収縮力が低下する。
(B) 収縮期のカルシウムイオン濃度の低下にもかかわらず、カルシニューリンなどのカルシウムイオン依存性シグナリングが活性化し病態を進行させる。心筋細胞内のように常にカルシウムイオン濃度が変化する状態で、カルシウムイオン依存性シグナリングが活性化するメカニズムは謎だった。
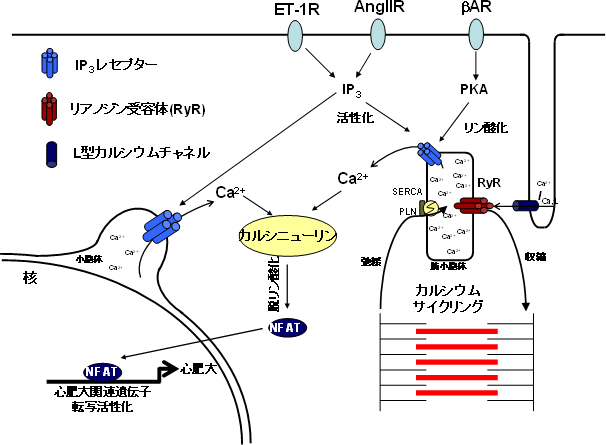
図2 IP3レセプターからのカルシウムイオン流出とカルシニューリンを介した心肥大
心臓の収縮と弛緩はカルシウムイオンサイクリングにより制御されている。その一方で、心臓の細胞膜上ではアンジオテンシンII 受容体(AngIIR)やエンドセリン-1(ET-1R)受容体刺激によりIP3 が生成し、IP3レセプターを活性化する。また、βアドレナリン受容体(βAR)刺激により活性化されるプロテインキナーゼA(PKA)が、IP3レセプターをリン酸化しIP3に対する感受性を増加させる。これらのメカニズムにより、心筋細胞においてIP3レセプターからカルシウムイオン流出が生じる。今回、生体心でIP3レセプターが、カルシウムサイクリングから独立してカルシニューリンを活性化し、NFATの脱リン酸化による核内移行を介して心肥大を引き起こすことが判明した。