理化学研究所(理研)生命機能科学研究センター分子機能シミュレーション研究チームの杉田有治チームリーダー(開拓研究本部杉田理論分子科学研究室主任研究員)、李秀栄上級研究員らの研究チームは、分子動力学(MD)計算[1]を用いて、酵素活性を低下させる阻害剤分子[2]が標的タンパク質に結合する際の複数経路と結合状態を特定し、結合初期に形成される複合体(会合体[3])が経路選択を制御していることを明らかにしました。
結合構造に基づいて設計された従来の薬剤分子には、標的タンパク質以外にも結合してしまうものがあり、副作用の原因になります。本研究成果は、結合経路の情報を用いることで、結合部位が似ているが機能の異なる標的タンパク質だけに結合するATP競合性阻害剤[2]などの設計に貢献すると期待できます。
今回、研究チームは、スーパーコンピュータ「京」[4]と高効率の構造探索アルゴリズムである「二次元レプリカ交換MD法(gREST/REUS法)[5]」を用いて、タンパク質リン酸化酵素の一つであるSrcキナーゼ[6]とATP競合性阻害剤間の結合・脱離イベントを100回程度サンプリングすることに成功しました。これまで、専用計算機を使ったMD計算で数回の結合イベントをサンプリングした例はありましたが、結合と脱離を同時に多数回サンプリングしたのは本研究が初めてです。これにより、結晶構造を水の位置も含めて高い精度で再現したほか、結晶構造では見えない準安定結合状態、およびそれらに至る複数経路と中間状態を高い信頼性で予測しました。そして、結合初期に形成される会合体の形と相互作用によって、異なる経路が選択されることを見いだしました。
本研究は、米国科学アカデミー紀要『Proceedings of the National Academy of Sciences of the United States of America(PNAS)』(8月26日号)に掲載されました。
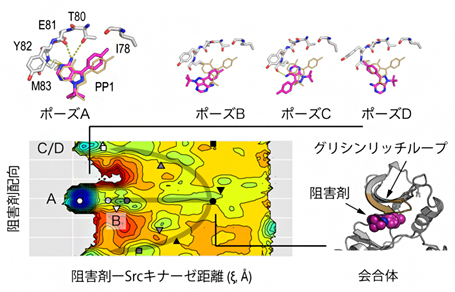
図 Srcキナーゼと阻害剤結合の複数経路と会合体の結合構造
背景
細胞機能の多くは、酵素(タンパク質)が基質分子と結合(分子認識)することで制御されています。例えば、細胞の外部から内部への信号伝達は、伝達経路に含まれるタンパク質がイオンや分子と結合し活性化することで行われます。タンパク質が基質分子を認識する機構を知ることができれば、細胞機能の理解が進み、効率の良い薬剤分子の設計が可能になります。
構造解析技術の進歩に伴い、基質分子と結合したタンパク質の高解像度X線結晶構造[7]や核磁気共鳴(NMR)構造[8]が豊富に得られるようになり、基質分子の認識と微細な結合様式との関係が明らかになってきました。しかし、これら従来の構造解析技術では、結合過程を直接観測するのは難しく、基質分子がどのような経路・状態を経てタンパク質に結合するのかは依然よく分かっていません。最近の薬剤分子設計は、標的タンパク質との結合親和性だけではなく、結合経路に関わるキネティクス[9](結合部位の滞在時間など)に基づいて行われていることから、結合過程の原子レベルでの理解が急務となっています。
分子動力学(MD)計算は、生体分子の動きを原子レベルで調べる有効な手段として、タンパク質、核酸や脂質といった生体分子の機能解析に広く用いられています。しかし、タンパク質と基質分子の結合は、ミリ秒かそれより長い時間スケールで起こるのに対して、現在MDで計算可能な追跡時間スケールはマイクロ(1マイクロ秒は1,000分の1ミリ秒)秒程度であるため、通常のMD計算で結合過程を調べるのは困難です。
MD専用スーパーコンピュータ[10]を用いると、数十マイクロ秒にわたる時間変化をシミュレーションでき、早い速度で起こる基質分子の結合であれば数回サンプリングすることが可能になっています。一方で、膨大な数の短い追跡時間の計算を行い、そのデータから長時間の分子運動を推定する手法や、基質分子の結合や脱離を促すバイアスポテンシャル[11]を課す手法などが開発されています。
研究手法と成果
研究チームは、タンパク質リン酸化酵素の一つであるSrcキナーゼとATP競合性阻害剤の結合に注目し、スーパーコンピュータ「京」と高効率の構造探索アルゴリズムであるレプリカ交換MD法[5]を用いて、阻害剤分子の結合・脱離を、より直接的に多数回サンプリングすることを考えました。レプリカ交換MD法の一種であるREST法では、異なる「溶質分子」の温度を持つ複数のレプリカを用意して、計算の途中でその温度を交換することにより、高温で広い構造空間を探索しつつ低温での安定構造を効率的に探索します。理研で開発しているMD計算ソフトウェアGENESIS(GENeralized Ensemble SImulation System)[12]に導入されているgREST法[5]では、REST法の「溶質分子」の定義を拡張して、「阻害剤分子とタンパク質結合部位の残基」の温度を変化させることができます。
今回、このgREST法とタンパク質-阻害剤分子の距離をパラメータとするREUS法を組み合わせた「二次元レプリカ交換MD法(gREST/REUS法[5])」を確立しました。そして、タンパク質リン酸化酵素の一つであるSrcキナーゼとATP競合性阻害剤の結合に注目した解析を行いました。今回開発した方法により、基質結合部位の構造ゆらぎが効果的に取り込まれた結果、結合と脱離に伴う活性化エネルギーを乗り越え、合計43マイクロ秒の追跡時間の計算で100回程度の結合・脱離イベントをサンプリングすることに成功しました。結合と脱離を同時に多数回サンプリングしたのは、本研究が初めてです。これにより、結晶構造を水の位置も含めて高い精度で再現したほか、結晶構造では見えない準安定結合状態[13]、およびそれらに至る複数経路と中間状態を、統計に基づいた高い信頼性で予測しました(図1)。
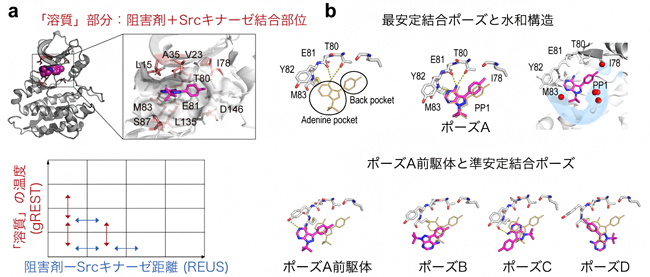
図1 二次元レプリカ交換MD法およびSrcキナーゼと阻害剤の結合構造
- a: 二次元レプリカ交換MD法(gREST/REUS法)のスキーム。「溶質」部分の温度(gREST)と阻害剤-Srcキナーゼ距離(REUS)のパラメータを交互に交換する。
- b: 上段は左から、結晶構造(黄色の棒表示)、今回予測された結合構造(最安定結合ポーズ)、その水和構造を示す。最安定結合ポーズでは、予測した水分布(水色)が結晶構造の水の位置(赤い球)と良く一致した。下段は、左からポーズAの前駆体と準安定状態の結合ポーズ(B、C、D)を示す。
計算データを解析した結果、阻害剤分子は結晶構造に見られる結合ポーズ(Aポーズ)以外に、配向の異なる三つのポーズ(Bポーズ、Cポーズ、Dポーズ)で結合できることを見いだしました。計算で得られた自由エネルギー地形[14]から、各結合ポーズは高いエネルギー障壁で隔たれるのに対して、各々の会合体はエネルギー的に行き来が可能であることが分かりました(図2上段)。このことから、各ポーズに至る経路は独立しており、それらは結合初期の会合状態でのみ交わるといえます。これは、結合初期に形成される会合体の形と相互作用によって異なる経路が選択されることを意味しています(図2下段)。会合体の構造を調べた結果、キナーゼの機能調節部位として知られるグリシンリッチループ[15]が、初期の阻害剤分子の捕捉とその後の経路選択を担っていることが分かりました。
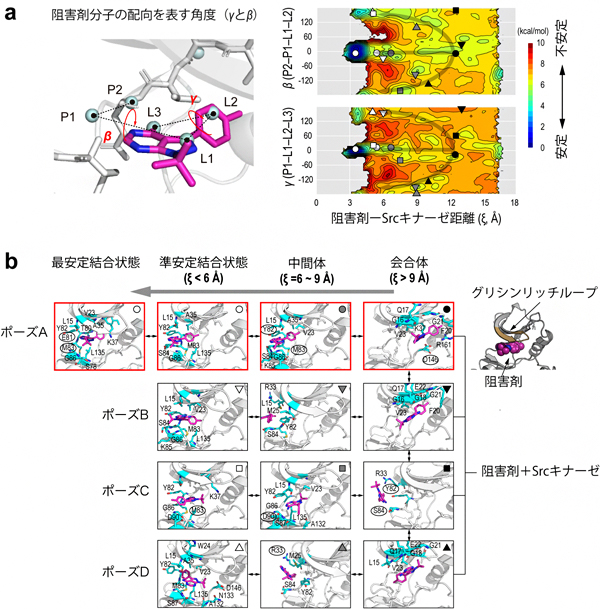
図2 Srcキナーゼと阻害剤分子の複数の結合経路
- a: 右側は、左側に示す阻害剤-Srcキナーゼ距離と阻害剤分子の配向を表す二つの二面角(βとγ)について求めた結合の自由エネルギー地形。クラスタリング解析で求めた代表構造(下段)の位置を地形に記号として示す。ポーズA~Dの各結合状態(○、▽、□、△)は、高いエネルギー障壁(赤で示される不安定な領域)で隔たれており、互いに行き来しにくい。これに対して、各々の会合体(●、▼、■、▲)の間には目立ったエネルギー障壁が見られず、エネルギー的に行き来しやすいことが分かる。
- b: 異なるポーズに至る経路と代表構造。右上部は、会合体におけるグリシンリッチループと阻害剤分子との相互作用。
これらの結果は、実験では見えない準安定結合ポーズや会合体の相互作用を調整することで、阻害剤分子の結合を制御できることを示しています。
今後の期待
本成果は、結合構造に基づいた従来の薬剤分子設計に対して、結合経路に基づいた新たな設計の可能性を示しています。今後、ブラウン動力学法[16]などと組み合わせれば、結合経路の詳細とともにキネティクスのパラメータである結合速度定数と解離速度定数の高精度予測も可能になります。
また、ATP競合性阻害剤の選択性向上は副作用の軽減に必須であり、本成果が理論・実験相補的な精密設計の新たな糸口となると期待できます。
さらに、本研究で確立したgREST/REUS法は、フリーソフトウェアであるGENESISに導入・公開されており、アカデミアのみならず産業界でも利用可能です。2021年以降に利用可能となる新しいスーパーコンピュータ「富岳」を用いた重要なアプリケーションの一つとなることも期待できます。
補足説明
- 1.分子動力学(MD)計算
原子間に働く力を計算し、運動方程式を繰り返し解くことで、分子の動きを追跡する方法。MDはMolecular Dynamicsの略。 - 2.阻害剤分子、ATP競合性阻害剤
阻害剤分子は、タンパク質酵素に結合して、酵素活性を低下または消滅させる分子のこと。ATP競合性阻害剤は、タンパク質リン酸化酵素のATP結合部位に競合的に結合し、その機能を阻害する分子のこと。 - 3.会合体
基質分子が標的タンパク質と出会い、非特異的な相互作用により過渡的に形成される複合体。 - 4.スーパーコンピュータ「京」
文部科学省が推進する「革新的ハイパフォーマンス・コンピューティング・インフラ(HPCI)の構築」プログラムの中核システムとして、理研と富士通が共同で開発を行い、2012年に共用を開始した計算速度10ペタフロップス級のスーパーコンピュータ。 - 5.二次元レプリカ交換MD法(gREST/REUS法)、レプリカ交換MD法
レプリカ交換MD法は、構造探索効率を上げるための方法の一つ。系の複数コピー(レプリカ)に対して温度の異なるMD計算を実施して、レプリカ間で温度を適宜交換することで構造探索の効率を上げる。REST法は、系の一部(溶質分子)の温度のみ異なるレプリカ間で交換を行うことで、少ないレプリカ数で構造探索の効率を上げる方法。Replica-Exchange with Solute Temperingの略。gREST法は、「溶質分子」の定義を拡張した方法。generalized RESTの略。REUS法は、多次元レプリカ交換法の一つで、温度以外のパラメータを交換する方法。Replica-Exchange Umbrella Samplingの略。上記gREST法とREUS法を組み合わせた二次元レプリカ交換MD法(gREST/REUS法)では、溶質温度と構造パラメータを交互に交換する。 - 6.Srcキナーゼ
Src遺伝子にコードされる非受容体型チロシンキナーゼタンパク質(他の分子のチロシン残基をリン酸化するタンパク質)。細胞成長の制御やがんと関係することが知られている。 - 7.X線結晶構造
タンパク質が規則正しく並んだ結晶にX線を照射し、得られた回折光を解析することでタンパク質の立体構造を解明するX線結晶構造解析により得られた構造。X線結晶構造解析では、タンパク質の立体構造を原子分解能で決定できる。 - 8.核磁気共鳴(NMR)構造
核磁気共鳴(NMR)法を用いて決定したタンパク質の立体構造。NMR法は、タンパク質を構成する原子の外部磁場への応答を利用した構造解析法で、溶液状態での解析ができ、構造情報に加え緩和時間など動的な情報を得ることができる。NMRはNuclear Magnetic Resonanceの略。 - 9.キネティクス
ここでは、化学反応の時間変化(反応速度)を意味する。 - 10.MD専用スーパーコンピュータ
分子動力学(MD)計算の高速化に特化して設計されたスーパーコンピュータ。代表的なものに、米国のANTONや日本のMDGRAPEがある。 - 11.バイアスポテンシャル
ここでは、シミュレートする分子システムをある状態に拘束、またはシフトするために(人為的に)付加するポテンシャルを指す。 - 12.GENESIS (GENeralized Ensemble SImulation System)
理研計算科学研究センターを中心に開発されている分子動力学ソフトウェア(GENESIS)。細胞環境を含む大規模な生体分子系のシミュレーションやレプリカ交換法などの構造探索手法を利用できるという特徴を持つ。 - 13.準安定結合状態
タンパク質-基質分子の結合状態のうち、安定性が低く短寿命で、X線結晶構造解析などでは捉えることが難しい結合状態。 - 14.自由エネルギー地形
分子システムのエネルギーを座標の関数として表したもの。通常は、特定の反応を表す座標(反応座標)に射影した地形を指す。分子運動や統計的な性質を視覚的に捉えることができる。 - 15.グリシンリッチループ
タンパク質キナーゼのヌクレオチド結合領域にある、グリシンを多く含んだ領域。GループやPループと呼ばれ、触媒作用に不可欠な領域として知られる。 - 16.ブラウン動力学法
溶媒分子を明示的に扱う代わりに、その効果をランダム力として計算に取り込んだ手法。シミュレーション系の原子数が少なく、長いタイムステップをとれる利点がある。
研究チーム
理化学研究所
生命機能科学研究センター 分子機能シミュレーション研究チーム
チームリーダー 杉田 有治(すぎた ゆうじ)
(開拓研究本 部杉田理論分子科学研究室 主任研究員
上級研究員 李 秀栄(り すよん)
研究員 尾嶋 拓(おしま ひらく)
研究員 笠原 健人(かさはら けんと)
計算科学研究機構(研究当時) 粒子系生物物理研究チーム
特別研究員(研究当時) 神谷 基司(かみや もとし)
研究支援
本研究の大部分の計算は、文部科学省ポスト「京」重点課題1「生体分子システムの機能制御による革新的創薬基盤の構築」におけるサブ課題A「ポスト『京』でのMD高度化とアルゴリズム深化(hpci170254など)」の一環として、「京」の計算資源を用いて行われました。
原論文情報
- Suyong Re, Hiraku Oshima, Kento Kasahara, Motoshi Kamiya, and Yuji Sugita, "Encounter Complexes and Hidden Poses of Kinase-Inhibitor Binding on the Free-Energy Landscape", Proceedings of the National Academy of Sciences of the United States of America, 10.1073/pnas.1904707116
発表者
理化学研究所
主任研究員研究室 杉田理論分子科学研究室
主任研究員 杉田 有治(すぎた ゆうじ)
生命機能科学研究センター 分子機能シミュレーション研究チーム
上級研究員 李 秀栄(り すよん)
 杉田 有治(左)李 秀栄(右)
杉田 有治(左)李 秀栄(右)
報道担当
理化学研究所 広報室 報道担当
Tel: 048-467-9272 / Fax: 048-462-4715
お問い合わせフォーム