Apr. 20, 2009 Press Release Biology
International FANTOM consortium publishes three milestone papers based on large-scale genome-wide data analysis
The international FANTOM consortium announces publication of three milestone papers in the prestigious journal Nature Genetics that will challenge current notions of how genes are controlled in mammals.
FANTOM, or Functional Annotation of the Mammalian cDNA, which is organized by RIKEN Omics Science Center (OSC), has leading scientists in Australia, Switzerland, Norway, South Africa, Sweden, Canada, Denmark, Italy, Germany, Singapore, UK, and the United States. The consortium has been providing the scientific community with extensive databases on the mammalian genome that describe molecular function, biology, and cell components.
FANTOM has become a world authority on the mammalian transcriptome, the set of all messenger RNA showing active genetic expression at one point in time. Other major discoveries are that approximately 70% of the genome is transcribed and that more than half of the expressed genes are likely non-coding RNAs (ncRNAs) that do not code proteins; thus, the prevailing theory that only 2% of the genome is transcribed into mRNA coding to proteins needed to be reexamined.
Now in its fourth stage, FANTOM4, led by OSC's Dr. Yoshihide Hayashizaki, has in over 3 years of laborious research developed a novel technology for producing a genome-wide promoter expression profile, established a mathematical scheme for describing the data obtained, and extracted key genomic elements that play dominant roles in the maintenance of cellular conditions.
In the current research, OSC has broadened its original technology CAGE (Cap Analysis of Gene Expression) and created deepCAGE, which takes advantage of next-generation sequencing to both precisely identify transcription start sites genome wide as well as to quantify the expression of each start site. The deepCAGE technology was applied to a differentiating acute myeloid leukemia cell line (ACL) to provide genome-wide time course dynamics of expression at the level of individual promoters - specific sequences on the DNA providing binding sites for RNA polymerase and the protein transcription factors that recruit them. The consortium built a quantitative model of the genome-wide gene expression dynamics that identified the key regulator motifs1 driving the differentiation, the time-dependent activities of the transcription regulators binding the motifs, and the genome-wide target promoters of each motif.
Validation of the model was performed by knocking down each transcription factor with small interfering RNAs. This first report of a large-scale gene network based on experimental data set is certain to generate much excitement in the scientific community. This information is also important for life science and medical researchers who are trying to uncover the processes by which cells undergo conversion or become cancerous, and for those attempting to determine how to control the growth and differentiation of stem cells and ensure their safety for use in regenerative medicine. Dr. Harukazu Suzuki, the scientific coordinator of the consortium, had this to say, "We are proud that we have created groundbreaking research in understanding more about how genes regulate cells at the molecular level and we want to acknowledge all consortium members for their great contribution to the research effort."
The FANTOM consortium has also expanded earlier discoveries of transcriptional complexity by exploring repetitive elements found throughout mammalian genomes with DeepCAGE. These elements, which constitute up to half of the genome, have been generally considered to be junk or parasitic DNA. However, the team has found that the repetitive elements are broadly expressed and 6 to 30% of mouse and human mRNAs are derived from repetitive element promoters. These RNAs are often tissue-specific and dynamically controlled, and control the output of the genome through a variety of mechanisms. The FANTOM4 collaborators have also identified yet another type of short RNA, referred to as tiRNA (transcription initiation RNA) or tiny RNAs, in the human, chicken, and Drosphilia. They are about 18 nucleotides (nt) in length and are found within -60 to +120 nt of transcription start sites and may actually be widespread in metazoans (animals). A BioMed Central Thematic Series features even more FANTOM 4 research papers in Genome Biology and several BMC journals.
1 Motif is a unique DNA sequence to which a corresponding group of transcription factor proteins binds.
Contact
Yoshihide Hayashizaki
Harukazu Suzuki
RIKEN Omics Science Center
Tel: +81-45-503-2222 / Fax: +81-45-503-9216
Jens Wilkinson
RIKEN Global Relations and Research Coordination Office
Tel: +81-(0)48-462-1225 / Fax: +81-(0)48-463-3687
Email: pr@riken.jp
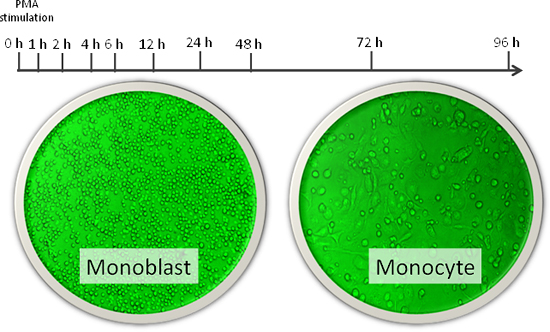
Figure 1
Cells used were human THP-1 cells (leukemia cells made to turn into monocytes using phorbol myristate acetate). Extensive data on transcription start sites was collected using OSC's originally developed deepCAGE technology during proliferation of monoblast into mature monocyte at ten time points (hours shown).
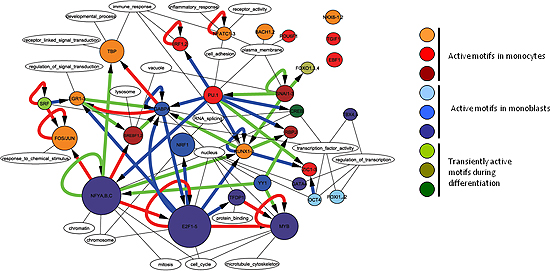
Figure 2
Transcription regulatory network predicted by FANTOM4 showing 30 key transcription factors (core motifs). Core network of 55 highly trusted edges made by filtering predicted edges from data in experiments validated by FANTOM or in the literature.