Oct. 25, 2010 Press Release Biology
Whole genome sequencing of Japanese individual reveals wealth of undiscovered genetic variation
Researchers at the RIKEN Center for Genomic Medicine (CGM) have uncovered hundreds of thousands of previously unknown variations in the human genome using new massively parallel sequencing technology. The findings, based on the complete sequencing of the genome of a single Japanese individual, provide vital clues on the role of rare genetic variants in disease susceptibility.
In recent years, advancements in DNA genotyping technologies have produced increasingly detailed information on the genetic variants, known as single nucleotide polymorphisms (SNPs), implicated in susceptibility to common diseases. Such technologies, however, target only common variants, whose influence on susceptibility is limited, leaving unaddressed the role of rare or novel variants.
Results of the research group's study, published in the journal Nature Genetics, mark a key step toward clarifying this role. Using massively parallel sequencing technology, one of the most powerful tools for discovering genome-wide variation, the group analyzed the complete genome of a Japanese individual, the first time this has ever been done.
Using a Bayesian decision method, the group identified over 3 million single nucleotide variations (SNVs), the most abundant and important type of variants in the human genome. Comparing these results to the genomes of six individuals from countries around the world reported in earlier studies, the group found numerous SNVs with an influence on gene function that had been previously overlooked. The group also identified 3 million base pairs of novel sequence not present in reference data from the Human Genome Project.
As the first whole genome sequencing of a Japanese individual, the results offer valuable insights on disease susceptibility among Japanese people. They also highlight the rich diversity still remaining in the human genome, and the power of whole genome sequencing as a means to discovering it.
Reference
- Akihiro Fujimoto, Hidewaki Nakagawa, Naoya Hosono, Kaoru Nakano, Tetsuo Abe, Keith A Boroevich, Masao Nagasaki, Rui Yamaguchi, Tetsuo Shibuya, Michiaki Kubo, Satoru Miyano, Yusuke Nakamura, and Tatsuhiko Tsunoda. Whole genome sequencing and comprehensive variant analysis of a Japanese individual using massively parallel sequencing. Nature Genetics (2010). DOI: 10.1038/ng.691
Contact
Tatsuhiko Tsunoda
Laboratory for Medical Informatics
RIKEN Center for Genomic Medicine
Tel/Fax: +81-(0)45-503-9556
Jens Wilkinson
RIKEN Global Relations and Research Coordination Office
Tel: +81-(0)48-462-1225 / Fax: +81-(0)48-463-3687
Email: pr@riken.jp
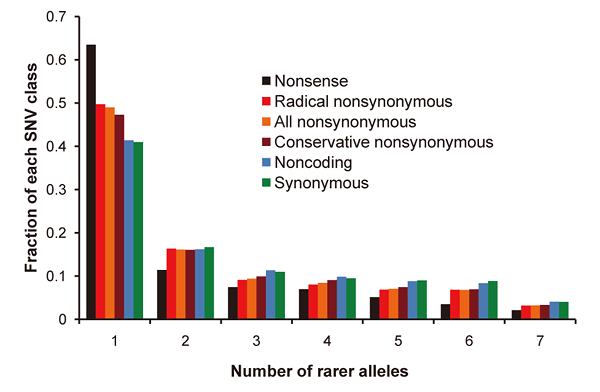
Figure 1: Allelic frequency spectrum of seven genomes
Many single occurrence alleles are observed that have yet to be fully explored. Also, because nonsense (black), radical nonsynonymous (red) and conservative nonsynonymous (brown) single nucleotide variants (SNVs) potentially have an impact on gene function, they are observed more as singletons in individuals (one allele) than are noncoding (blue) and synonymous (green) SNVs.
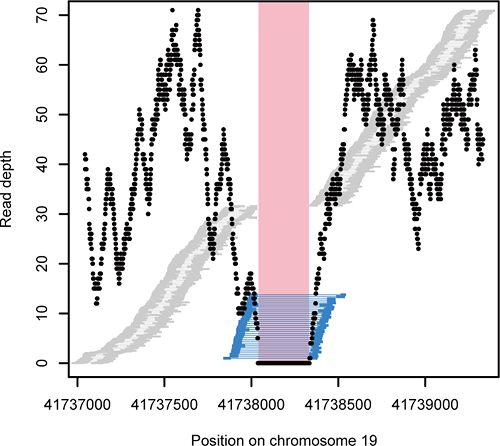
Figure 2: Deletion detection by mapping distance and read depth
Candidate deletions in the genome were identified by the minimum overlap region between read pairs that map further apart than expected (blue bars). The read depth (black dot) of this region was compared to that of the flanking regions.