Oct. 3, 2013 Press Release Biology Medicine / Disease
Advanced gene expression analysis to facilitate drug development
When developing new drugs, monitoring cellular responses to candidate compounds is essential for assessing their efficacy and safety. Researchers from the RIKEN Center for Life Science Technologies report a new method to monitor and quantify the activity of gene promoters during the response to a drug, using the advanced gene expression analysis method CAGE followed by single-molecule sequencing. This research paves the way to a more precise analysis of cellular responses to drugs, at the level of individual promoters.
The study is published this week in this week in the journal CPT: Pharmacometrics & Systems Pharmacology.
Microarray-based technologies are widely used to monitor cellular changes in response to drug administration at the level of genes. However, microarrays have several limitations due to the fact that they rely on pre-designed oligonucleotide probes and detection based on hybridization.
In order to circumvent the limitations imposed by the use of microarray-based technology for the development of new drugs, Dr Harukazu Suzuki and his team at CLST developed a new technique combining Cap Analysis of Gene Expression (CAGE) with 3rd generation, single-molecule sequencing.
CAGE is a method developed at RIKEN to comprehensively map human transcription start sites and their promoters and quantify the set of mRNAs in a cell, also called the transcriptome.
During CAGE, the 5’-end of mRNAs is sequenced in order to produce a series of 20-30 nucleotide sequences that can then be mapped onto the genome and provide information about the level of expression of genes.
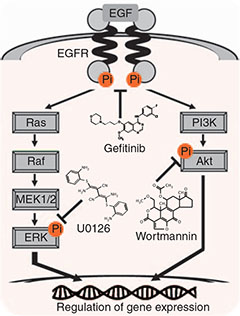
Dr Suzuki and his team used CAGE, combined with a single-molecule sequencer, to monitor the effect of three drugs, U0126, wortmannin and gefitinib on human breast cancer cells.
U0126 and wortmannin are known to inhibit the Ras-ERK and phosphatidylinositol-3-kinase (PI3K)-Akt signalling pathways within cells. Gefinitib is a potent inhibitor of the epidermal growth factor receptor kinase (EGFR kinase) and mainly inhibits the Ras-ERK and PI3K-Akt pathways downstream of EGFR.
The researchers identified a distinct set of promoters that were affected by low doses of the drugs, and therefore showed sensitivity to a weak inhibition of the Ras-ERK and PI3K-Akt signal-transduction pathways. This level of precision would would have been very difficult to achieve using microarray-based profiling.
Furthermore, a quantitative analysis showed that the inhibitory profiles of both U0126 and wortmannin are constitutive components of the transcriptome profile obtained by inhibition of the EGFR kinase. Using a regression model, the researchers were able to quantitatively predict the promoter activity profile of gefitinib, based on the U0126 and wortmannin profiles.
These results demonstrate the potential utility of highly quantitative promoter activity profiling in drug research.
“Quantitative transcriptome analysis is potentially widely applicable to determine the target proteins and action mechanisms of uncharacterized compounds,” concludes Dr Suzuki. “Our study paves the way for quantitative analysis of drug responses at the promoter level, and moreover, is potentially applicable for the evaluation of combinatorial or serial drug treatment in a clinical setting,” he adds.
Reference
- Kazuhiro Kajiyama, Mariko Okada-Hatakeyama, Yoshihide Hayashizaki, Hideya Kawaji, Harukazu Suzuki. "Capturing drug responses by quantitative promoter activity profiling." CPT Pharmacometrics and Systems Pharmacology, 2013, doi: 10.1038/psp.2013.53
Contact
Harukazu Suzuki, Group Director
Omics Application Technology Group
Division of Genomic Technologies
RIKEN Center for Life Science Technologies
Jens Wilkinson
RIKEN Global Relations and Research Coordination Office
Tel: +81-(0)48-462-1225 / Fax: +81-(0)48-463-3687
Email: pr@riken.jp
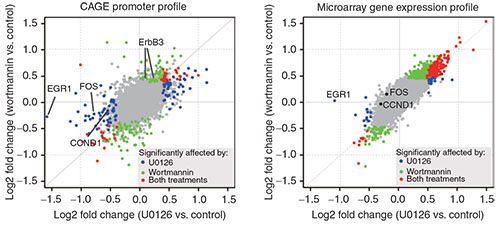
Promoter-based expression profiling of drugs targeting the epidermal growth factor receptor (EGFR) pathway