Oct. 21, 2014 Press Release Biology Medicine / Disease
Scientists unravel the mystery of a rare sweating disorder
An international research team discovered that mutation of a single gene blocks sweat production, a dangerous condition due to an increased risk of hyperthermia, also known as heatstroke. The gene, ITPR2, controls a basic cellular process in sweat glands, promoting the release of calcium necessary for normal sweat production, and its loss results in impaired sweat secretion.
The study, published on October 20, in the Journal of Clinical Investigation, was led by Katsuhiko Mikoshiba, a molecular cell biologist from the RIKEN Brain Science Institute in Japan and Niklas Dahl, a genetics expert at Uppsala University in Sweden.
Dr. Dahl's group studies rare single-gene diseases, also known as Mendelian disorders, with the goal of identifying causative genes to develop diagnostic or treatment tools. Working with collaborators in Pakistan, they identified a family with several children who could not sweat, a rare condition called anhidrosis.
Sweating serves to regulate body temperature and keeps animals from overheating. Although excessive sweating is an embarrassing problem, an inability to sweat is potentially lethal, as it impairs the body's ability to regulate temperature and increases susceptibility to heatstroke.
Anhidrosis can have several causes, including genetic, in which case the sweat glands are either deformed or missing. In contrast, the sweat glands of the individuals examined in this study appeared visually normal and no other physical problems were reported.
Analysis of the patients' genomes revealed a mutation in a gene called ITPR2, which encodes IP3 receptor type 2 (IP3R2), a protein that forms a channel in the membrane of the endoplasmic reticulum, an organelle within cells that stores an ion called calcium. Opening of the IP3R channel releases calcium, triggering essential cell behaviors such as movement, shape changes, or secretion.
In order to understand how the mutation caused a lack of sweat production, Dr. Dahl sought collaboration with Dr. Mikoshiba, who discovered the IP3 receptor. "This is the first report of IP3 receptor type 2 mutation in human disease," said Dr. Mikoshiba. "The surprise was that a point mutation, not a large deletion was enough to cause the human disorder."
Dr. Mikoshiba's group examined the function of the mutated IP3R2 protein in cultured cells and found that a single nucleotide change in the DNA code resulted in a change in protein structure impeding the ability of the channel to release calcium. The team then examined sweat production in mice with a genetic loss of the IP3R2 protein and found that they had reduced sweating.
Calcium release is a potent signal within cells and some animals have several IP3 receptor types to compensate for each other if one is not functioning properly. The researchers found that human sweat glands mainly have IP3R2. However, in mouse, sweat glands also have R1 that may partially make up for the lack of R2, explaining why the mutant mice were still able to produce some sweat.
"Although anhidrosis is quite a rare condition, the 'opposite' phenotype, excess sweating or hyperhidrosis, is a common problem affecting 2% of the population" said Dr. Dahl. "Such symptoms may be alleviated by a drug that inhibits IP3R2. However," he cautions, "IP3R2 is expressed in many tissues, and compounds must first be carefully evaluated in experimental models." Efforts to develop drugs to regulate the IP3R are currently underway.
Reference
- Joakim Klar, Chihiro Hisatsune, Shahid M. Baig, Muhammad Tariq, Anna C.V. Johansson, Mahmood Rasool, Naveed Altaf Malik, Adam Ameur, Kotomi Sugiura, Lars Feuk, Katsuhiko Mikoshiba, and Niklas Dahl. "Abolished InsP3R2 function inhibits sweat secretion in both humans and mice". The Journal of Clinical Investigation, 2014, doi: 10.1172/JCI70720
Contact
Laboratory Head
Katsuhiko Mikoshiba
Laboratory for Developmental Neurobiology
RIKEN Brain Science Institute
Jens Wilkinson
RIKEN Global Relations and Research Coordination Office
Tel: +81-(0)48-462-1225 / Fax: +81-(0)48-463-3687
Email: pr@riken.jp
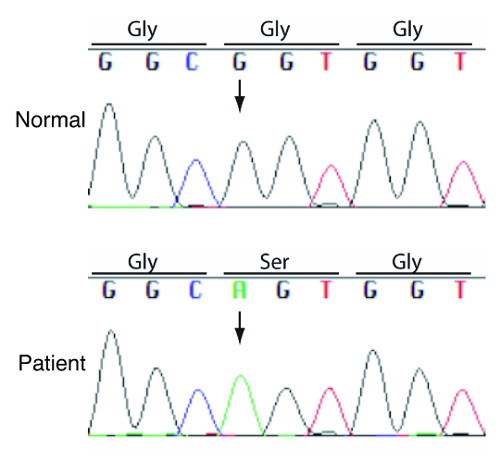
A single change in the DNA of the IPTR2 gene (arrow) changes the structure of the IP3R2 protein.
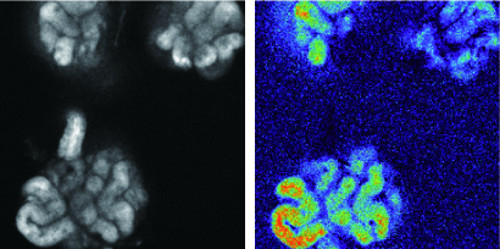
Image showing how calcium is involved in sweat production in a mouse sweat gland