Dec. 21, 2007 Research Highlight Biology
Poor transport cuts to the bone
Researchers link skeletal disorders with sugar chain production
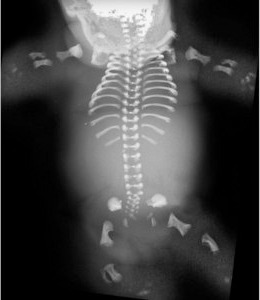 Figure 1: A human skeleton affected by Schneckenbecken dysplasia. © (2007) Peter Nikkels
Figure 1: A human skeleton affected by Schneckenbecken dysplasia. © (2007) Peter Nikkels
A international research team with strong RIKEN representation has uncovered a molecular basis—and developed a mouse model—for some types of congenital human skeletal disorders, including the rare, lethal Schneckenbecken dysplasia (Fig. 1). The team, led by Shiro Ikegawa of RIKEN’s SNP Research Center in Tokyo, has accumulated persuasive evidence that the underlying problem is an inoperative form of the molecule responsible for transporting sugars—critical to the construction of the skeleton—across the membrane of the cellular organelle known as endoplasmic reticulum (ER).
The skeleton is formed from a matrix of compounds secreted by cartilage and bone cells. A family of large molecules called proteoglycan is intimately involved in the process. The proteoglycan molecules consist of a protein core to which long chains of sugars, such as chondroitin sulfate, are attached. Chondroitin sulfate is made in the ER and another cellular organelle, known as the Golgi apparatus, from two different sugar compounds. Its composition reflects, and may even control, the growth activity of the bone. The sugars are moved into the ER by molecules known as nucleotide-sugar transporters (NSTs). More than 10 genes for NSTs are known in humans.
In a recent Nature Medicine paper1, researchers from Japan, the US, Germany and Holland, detailed how they developed a line of mice incorporating a recessive mutation in SLC35D1, the gene coding for the NST involved in chondroitin sulfate synthesis. They confirmed that mice in which this mutation is expressed died as newborns, and displayed skeletal abnormalities, in particular short, thick limbs. The researchers then used cell staining and radioactive labeling to confirm that, at a molecular level, these developmental problems were associated with a severe reduction in the length and number of sugar chains of the proteoglycans.
The mutant mice bore many features in common with humans suffering from Schneckenbecken (‘snail pelvis’) dysplasia. When the team investigated two human cases of this dysplasia, they were able to show that the NSTs involved in chondroitin sulfate synthesis were inactive. They were also able to trace the problem back to mutations in the human SLC35D1 gene.
This is the first demonstration of the consequences in humans of such abnormal biosynthesis of proteoglycans sugar chain. Examples of other human disorders associated with proteoglycans have recently been discovered. “We expect that our results will open many avenues of research in the fields of connective tissue disorders, vertebrate development and glycobiology,” Ikegawa says.
References
- 1. Hiraoka, S., Furuichi, T., Nishimura, G., Shibata, S., Yanagishita, M., Rimoin, D.L., Superti-Furga, A., Nikkels, P.G., Ogawa, M., Katsuyama, K., Toyoda, H., Kinoshita-Toyoda, A., Ishida, N., Isono, K., Sanai, Y., Cohn, D.H., Koseki, H. & Ikegawa, S. Nucleotide-sugar transporter SLC35D1 is critical to chondroitin sulfate synthesis in cartilage and skeletal development in mouse and human. Nature Medicine 13, 1363–1367 (2007). doi: 10.1038/nm1655