May 23, 2008 Research Highlight Biology
New potential to curb a neurodegenerative disease
Researchers pinpoint viable cellular targets for therapies to slow the progression of amyotrophic lateral sclerosis
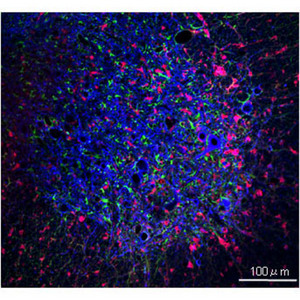 Figure 1: Labeled neuronal (blue), microglial (red), and astrocytic (green) cells in a spinal cord lesion of a mouse with ALS. Modified from Ref. 2 © 2008 Nature Publishing Group
Figure 1: Labeled neuronal (blue), microglial (red), and astrocytic (green) cells in a spinal cord lesion of a mouse with ALS. Modified from Ref. 2 © 2008 Nature Publishing Group
Amyotrophic lateral sclerosis (ALS or Lou Gehrig’s disease) is a neurodegenerative disease marked by a progressive, and eventually fatal, loss of motor neurons. Familial forms of the disease are associated with dominant mutations in the gene for the protein superoxide dismutase (SOD1). These mutations result in misfolding and aggregation of SOD1. The misfolded protein, in turn, causes motor neuron toxicity, though the mechanism for this effect is unknown.
ALS has been modeled in the mouse by expressing SOD1 harboring a human mutation. However, it was not clear whether mutant SOD1 expression in non-neuronal cells in the central nervous system also plays a role in ALS; namely, in microglia, resident immune cells responsible for neuro-inflammation, and in astrocytes, a supporting cell type that plays a principal role in brain repair (Fig. 1).
To address this question, Koji Yamanaka at the RIKEN Brain Science Institute in Wako, along with colleagues in Japan and the United States, developed a new mouse model of ALS that expresses a mutant version of the human SOD1 gene, but in such a way that the human gene can be deleted in specific cell types at will. When the team deleted the mutant human SOD1 in motor neurons, a delay in disease onset was observed1; whereas, deletion in either microglia1 or in astrocytes2 did not delay the onset but it did slow the rate of progression. Thus, as compared to controls, the ALS mice with deletion of the human mutation in microglia survived for an average of 99 days longer, while mice with the deletion in astrocytes survived for an average of 60 days longer.
The team’s findings suggest that onset of disease is due to motor neuron damage, but this damage in turn leads to a strong inflammation response by the microglia that further exacerbates the disease process. However, the team has found that mutant SOD1 damage in the astrocytes resulted in more of the microglial-derived inflammation, which may then damage the motor neurons further and thus astrocytes also play a role in late disease progression.
How the mutant astrocytes are affecting the neuro-inflammation response of the mutant microglia, however, remains unclear. Yamanaka plans to uncover this mechanism in a future study. And once identified he hopes that “the information could help target a therapy that can halt the inflammation response mediated by the astrocytes and microglia.” This would ameliorate ALS by slowing disease progression, even if disease onset due to motor neuron dysfunction is not preventable, he says.
References
- 1. Boillée, S., Yamanaka, K., Lobsiger, C.S., Copeland, N.G., Jenkins, N.A., Kassiotis, G., Kollias, G. & Cleveland, D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392 (2006). doi: 10.1126/science.1123511
- 2. Yamanaka, K., Chun, S.J., Boillée, S., Fujimori-Tonou, N., Yamashita, H., Gutmann, D.H., Takahashi, R., Misawa, H. & Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nature Neuroscience 11, 251–253 (2008). doi: 10.1038/nn2047