2012年2月13日
独立行政法人 理化学研究所
次世代シーケンサーのデータ解析精度を向上させる新手法を開発
-疾患を未然に防ぐ先制医療、バイオマス資源開発などへの応用に期待-
ポイント
- 断片化RNA情報から全長RNA情報を92.6%の高い成功率で再構築可能
- サンプル間のRNA分子配列の変化を検出し、変化の定量化が可能に
- 急速に進むデータの蓄積を解析精度向上に結びつける新たな手法
要旨
独立行政法人理化学研究所(野依良治理事長)は、次世代シーケンサー(超高速塩基配列解読装置)で得られた断片化RNA※1の情報から、細胞内に本来存在している全長RNA※1の情報を高精度に再構築する解析プログラム「ARTADE2(アートエイド ツー)」を開発しました。これは、理研生命情報基盤研究部門(理研BASE)の豊田哲郎部門長らによる研究成果で、「ARTADE2」のアプリケーションは理研BASEのwebページに掲載、2012年2月13日からダウンロードできます。
細胞内に存在するRNAのセット「トランスクリプトーム※2」は、細胞の機能を決定付ける重要な要素です。トランスクリプトームを正確に捉えることは、疾患を未然に防ぐ先制医療やバイオマス資源開発など、生命・環境科学分野でのさまざまな応用展開に有効です。近年の次世代シーケンサーの発展は、トランスクリプトームの観察に飛躍的な大規模化・効率化をもたらしましたが、入手できるRNA分子の塩基配列情報は、100塩基程度という短い断片化RNAに限られていました。そのため、断片化RNAの配列情報を正確に再構築し全長RNAの情報を得るための処理が必要であり、これを高い精度で実現する手法やソフトウェアなどの開発が求められていました。
理研BASEは、複数のサンプルから取得したmRNA-seq※3データを用いて、ゲノム上での転写活性の相関関係を解析する「ポジショナル相関解析法※4」を考案し、それを実行するプログラム「ARTADE2」を開発しました。実際にRNA構造が既知であるシロイヌナズナでARTADE2の精度を検証した結果、全長RNAの塩基配列情報の再構築を92.6%という高い成功率で実現しました。さらにこの手法を応用し、サンプル間のRNA分子の配列変化の検出や定量化にも成功しました。ARTADE2は、複数サンプルでの転写の相関情報を利用するため、サンプル数が増加していくにつれ、解析精度がより向上していきます。このため、近年急速に進んでいる次世代シーケンサーが生産するデータの蓄積を最大限に活用できる解析手法です。
本手法の応用として、健常者と疾患者のトランスクリプトームを比較した分子マーカーの同定による先制医療への貢献や、さまざまな植物のトランスクリプトームやその動態解析によるバイオマス資源開発などが計画されています。
本研究成果は、文部科学省 革新的細胞解析研究プログラム(セルイノベーション)、科学技術振興機構 統合化推進プログラムの助成を得て行われ、英国の科学雑誌『Bioinformatics』オンライン版(2月13日付け)に掲載される予定です。
背景
細胞内に存在するRNAは、細胞だけでなく生体全体の機能に大きな影響を及ぼします。RNAの状態は疾患との関連も深く、がんなどの疾患では特徴的なRNAの発現パターンや選択的スプライシング※5が生じることが知られています。また、この変化は疾患に先駆けて生じると考えられており、疾患の予兆となるような変化を探すことで疾患を未然に防ぐ「先制医療」へとつなげていくことが期待されています。
近年、次世代シーケンサーで細胞内のmRNAの状況を調べる手法「mRNA-seq」という手法が広く利用されています。次世代シーケンサーによるデータ生産量は年々増加しており、1回の実験で6,000億塩基(Illumina社Hiseq2000の場合)で、これはヒトゲノムDNAの200倍の量に相当します。一方、mRNAの長さはさまざまで、ヒトの場合、1,000塩基を超えるものがRNA全種類のうち80%以上を占めます。しかし、mRNA-seqでは、100塩基程度までの短い断片の情報しか読み取ることができません。このためRNAの状況を知るには、次世代シーケンサーで不完全に読み取られた断片化RNAの情報から完全な全長RNAの情報を再構築する処理が必要です。細胞内のmRNAの状態が転写やスプライシングの制御によって多様に変化することや、シーケンスの際に生じるノイズや偏りの存在がこの処理を困難にしていました。そこで、全長RNAの情報を高精度に再構築するためのプログラムが必要とされていました。
研究手法と成果
従来のソフトウェアは、一つのmRNA-seqデータだけを用いて処理しているため、データによるノイズや偏りの影響を大きく受けていました。理研BASEが開発した「ARTADE2」では、さまざまな生体組織や環境条件下で採取したRNAから複数のmRNA-seqデータを取得・統合して解析します。これによりノイズや偏りの影響の問題を解決しました。具体的には、同一のゲノムにおける異なる2点間のRNA発現活性の相関を調べる「ポジショナル相関解析」を網羅的に計算し、ゲノム配列情報と合わせて統合的に解析することで、全長RNAの情報を高い精度で再構築することに成功しました(図1)。
ARTADE2の性能を評価するため、理研植物科学研究センター(篠崎一雄センター長)の協力の下、シロイヌナズナから取得したRNAを用いてmRNA-seqを行い、このデータに対してARTADE2を適用しました。その結果、予測したRNA全長構造の92.6%を、既知のRNA構造に対して正しく再構築することに成功しました。この結果は、従来よく利用されている既存ソフトウェア(Cufflinks(カフリンクス):米メリーランド大学が開発)の78.6%を上回る結果となりました(図2)。
また、トランスクリプトーム解析に用いられるゲノムタイリングアレイ※6の結果にも、シロイヌナズナのデータに対してARTADE2を適用しました。その結果、1,000個以上の新規遺伝子やアンチセンスRNA※7を新たに発見しました。これは、ゲノム配列の意味、意義の正確な理解を、一層すすめていくことにつながります。
さらに、ポジショナル相関解析に多変量解析の1つである因子分析手法を組み合わせることで、転写開始点の変化や選択的スプライシングによって生じるRNAの塩基配列の変化を同定し、それぞれのサンプルにおける変化を定量化する解析手法も開発しました(図3)。この変化は、そこから翻訳されるタンパク質の配列やタンパク質への翻訳効率に変化をもたらします。また、RNA分子そのものが機能分子として振る舞う例も多く知られているため、トランスクリプトームの変化は、ゲノムの情報と細胞の表現形質をつなぐ重要な要素です。ARTADE2はゲノム、トランスクリプトーム、プロテオーム(細胞中のタンパク質全体)、フェノーム(細胞・個体の持つ表現形質の総体)という複数階層のオミックス研究を仲介する重要な技術となります。
ARTADE2は、複数のサンプルから得られたトランスクリプトームのデータからゲノム上の位置関係で転写活性の相関性を解析するため、サンプル数が増えるほどデータのノイズや偏りに対して頑強になり、解析精度が向上します。従って、近年急速に蓄積が進む次世代シーケンサーデータを有効活用することが可能です。
今後の期待
今後、ARTADE2を用いて、健常者と疾患者のRNAデータを比較・解析することにより疾患の分子メカニズムの解明につながると期待されます。また、ARTADE2で疾患の予兆となるようなRNAの変化を探し、これを分子マーカーとして用いることで疾患を発見できる可能性があります。これは疾患を未然に防ぐ「先制医療」へとつながります。
また、バイオ燃料やバイオプラスチックなどのバイオマス資源開発を目指すグリーンイノベーションでは、植物細胞内のRNAの構造や動態の理解は重要です。植物は多様性に富み、目的とするバイオマスによって利用に適した植物が異なります。研究対象となることの少ない非モデル植物ではトランスクリプトームの解析は困難ですが、ARTADE2を用いることで効率的かつ正確にトランスクリプトームの構造やRNAの動態を捉えることが可能になり、グリーンイノベーションへの貢献が期待できます。
原論文情報
- Shuji Kawaguchi, Kei Iida, Erimi Harada, Kousuke Hanada, Akihiro Matsui, Masanori Okamoto, Kazuo Shinozaki, Motoaki Seki, and Tetsuro Toyoda
“Positional correlation analysis improves reconstruction of full-length transcripts and alternative isoforms from noisy array signals or short reads”.
Bioinformatics,2012,doi:10.1093/bioinformatics/BTS065
発表者
理化学研究所
生命情報基盤研究部門
部門長 豊田 哲郎(とよだ てつろう)
Tel: 045-503-9610 / Fax: 045-503-9553
お問い合わせ先
横浜研究推進部 企画課
Tel: 045-503-9117 / Fax: 045-503-9113
報道担当
理化学研究所 広報室 報道担当
Tel: 048-467-9272 / Fax: 048-462-4715
補足説明
- 1.断片化RNA、全長RNA
RNA分子はDNA分子と同様、4種類のヌクレオチドが直鎖状に重合している分子である。メッセンジャーRNA (mRNA)と呼ばれるタイプのRNA分子はゲノムDNAを鋳型として合成され、ゲノム情報を仲介する分子としてふるまう。このため細胞内のRNA分子におけるヌクレオチドの順番や重合数(あわせてRNAの配列と呼ぶ)が重要になる。RNA分子にはさまざまな長さのものが存在し、例えばヒトにおいては、RNA分子全種類の80%以上のものは1,000ヌクレオチドを超える長さを持っていると考えられている。現在、一般に用いられている次世代シーケンサーを使ったmRNAの解読手法(mRNA-seq、 ※3参照)を用いるとmRNA分子の配列は、解析の際の操作やシーケンサーの技術的な問題により、100塩基以下の短い分子の情報として解読される。この短くなったRNA分子をここでは断片化RNAと呼び、対比して、元来のRNAを全長RNAと呼ぶ。 - 2.トランスクリプトーム
ゲノムDNAを鋳型としてRNA分子が合成されることを転写(トランスクリプション)と呼び、転写の結果合成されたRNA分子のことを転写産物と呼ぶ。細胞中にはさまざまなRNA分子がそれぞれの量で存在している。このようなRNA分子の総体を、転写産物を表す「トランスクリプト」に総体を表す接尾語「オーム」をつなげて「トランスクリプトーム」と呼ぶ。 - 3.mRNA-seq
ゲノムから転写されるRNAのうちタンパク質情報をコードしているRNAのことをmRNAと呼ぶ(*1も参照)。mRNA-seqは、次世代シーケンサーを用いてmRNA分子の配列を解読する実験手法名。 - 4.ポジショナル相関解析法
次世代シーケンサーによる複数個のトランスクリプトーム観察結果を用い、ゲノム上の異なる2点間のRNA転写活性の相関を網羅的に調べることで、細胞内のRNA分子の手がかりを得る解析手法。理研BASEが開発した手法で、複数のトランスクリプトームデータを統合し、高精度な解析を可能とする世界で初めての手法。 - 5.選択的スプライシング
真核生物のmRNAの多くは、ゲノムDNAから転写を受けた後、アミノ酸配列情報を持たない部分(イントロン)が切り出され、アミノ酸配列情報を持つ部分(エキソン)がつなぎ合わされることで成熟mRNAとなる(スプライシング)。 mRNAによってはエキソンとイントロンの位置が一定ではなく、1種類の未成熟mRNAから複数種類の成熟mRNAが作成される場合がある。この現象を選択的スプライシングと呼ぶ。 - 6.ゲノムタイリングアレイ
トランスクリプトーム解析に使われる解析ツールの1つ。あるゲノム領域から転写されるRNAに対応した相補鎖が用意されており、そのRNAが逆転写したDNA(ラベル化したcDNAを用いることで、網羅的にゲノムの転写活性を測定することができる。 - 7.アンチセンスRNA
ゲノムを構成する二本鎖DNAにおいて、片方の鎖から転写されるRNA(センスRNA)に対して、逆の鎖から転写されるRNAをアンチセンスRNAと呼ぶ。センスRNAに対して阻害したり、調節したりする役割を持つことが知られている。
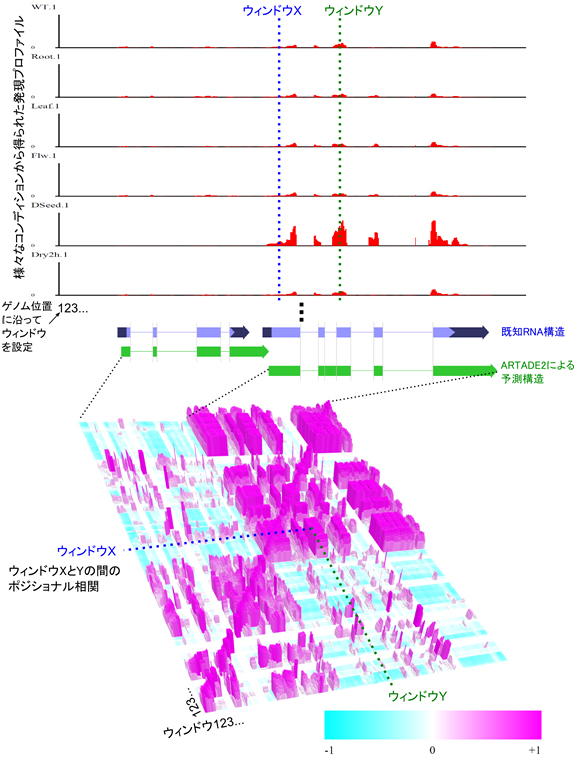
図1 複数個のmRNA-seqデータに対してポジショナル相関解析を適用した結果
- 上段:個々のmRNA-seqデータ。縦軸はRNA発現の強さを、横軸はゲノムの位置を示す。図中のX-Yはポジショナル相関計算の例(下段)と対応している。
- 下段:ポジショナル相関解析結果。横・縦軸は共にゲノムの位置を示しており上段図と対応している。ポジショナル相関が高いほど赤く、高い四角柱で表示されている。トランスクリプトーム観察結果には多くのノイズが含まれるが、ポジショナル相関解析を行うことで、RNA分子のエキソンに対応する領域が、高いポジショナル相関を示す「島(右上と左下にある赤色の四角形)」として可視化される。ARTADE2はこの情報を利用し、RNA分子の全長構造を再構築する。
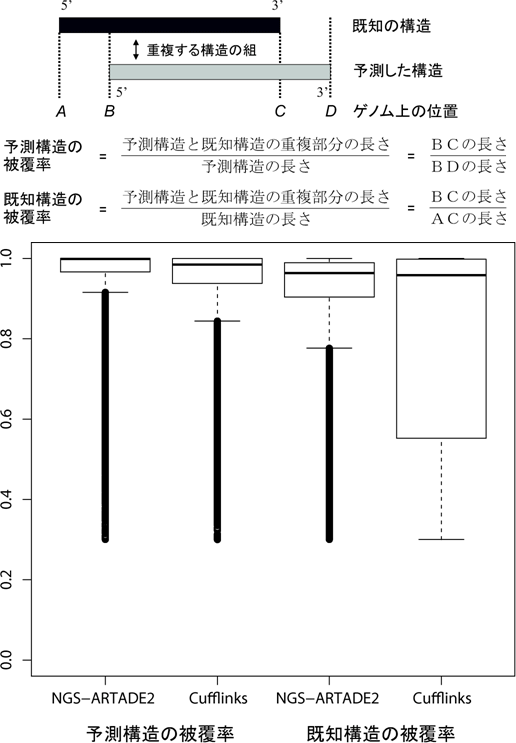
図2 ARTADE2結果の精度検証結果
ARTADE2(左から1番目、3番目)とCufflinks(2番目、4番目)それぞれの結果を既知RNA全長構造と比較し、縦軸に被覆率を示した。ここでは箱が上に表示されるほど、予測構造と既知構造が一致していることを示す。
- 予測構造の被覆率:左から1、2番目は、予測結果に対する被覆率の分布を示す。予測したRNAの長さのうち、どれだけの長さが既知構造と対応していたかを計算してその分布をプロットした。
- 既知構造の被覆率:左から3、4番目は、既知構造に対する被覆率の分布を示す。既知のRNAの長さのうち、どれだけの長さが予測構造と対応しているかを計算してその分布をプロットしている。この分布の比較により、全長構築の成功率はARTADE2(3番目)がCufflinksの結果(4番目)よりも優れていることが分かった。文中の全長構造の成功率の計算では、既知RNA構造の80%以上の領域を予測できている場合を成功とした。
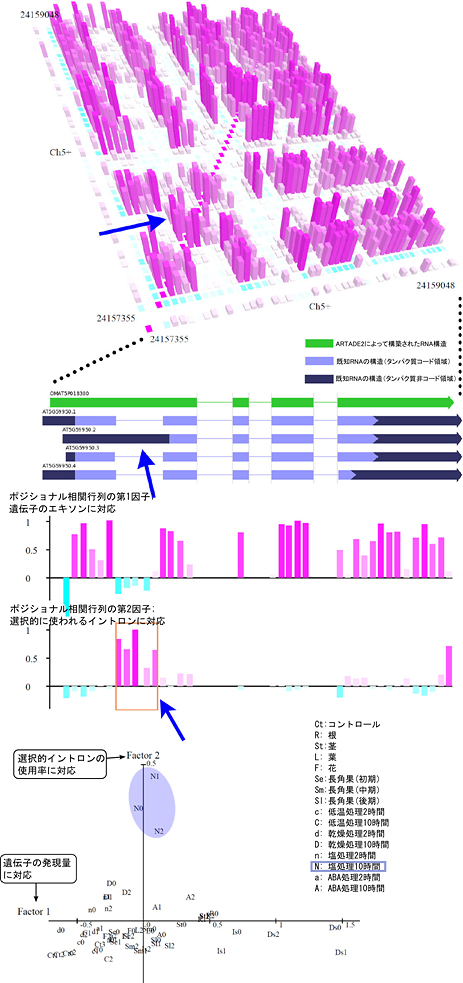
図3 因子分析手法による選択的スプライシング領域の同定と変化の定量化
- 上段:選択的スプライシングが生じるRNAのポジショナル相関解析例。青矢印が選択的スプライシングによるイントロンの変化。この影響を受ける領域は、領域内での相関は高いがRNAの他の領域との相関は低い特徴を持つ。
- 中段:ARTADE2によるRNA構造の再構築結果と、対応する既知遺伝子のエキソン構造、因子分析による選択的スプライシング領域の同定結果。青矢印は上段で示す領域と対応。ポジショナル相関解析で示される選択的スプライシング領域は既知のそれと対応し、因子分析によりこの領域をポジショナル相関行列の第2因子として同定することを実現した。
- 下段:因子分析結果による、各解析サンプルの定量化結果。選択的スプライシング領域を同定すると同時に、各サンプルのRNA構造の変化を定量化できる。この場合、塩処理10時間後のサンプルで、選択的イントロンの使用率が上昇することを示す。