要旨
理化学研究所(理研)統合生命医科学研究センターゲノムシーケンス解析研究チームの中川英刀チームリーダー、藤本明洋副チームリーダー、国立がん研究センター がんゲノミクス研究分野 柴田龍弘分野長、十時泰ユニット長、東京大学医科学研究所附属ヒトゲノム解析センターの宮野悟教授、広島大学大学院医歯薬保健学研究院の茶山一彰教授らの共同研究グループ※は、日本人300例の肝臓がんの全ゲノムシーケンス解析[1]を実施し、それらのゲノム情報を全て解読しました。この研究は、国際がんゲノムコンソーシアム(ICGC)[2]のプロジェクトの一環として行われ、単独のがん種の全ゲノムシーケンス解析数としては世界最大規模となりました。
日本では、年間約4万人が肝臓がんと診断され、3万人以上が亡くなっています。特に、日本を含むアジアで発症頻度が高く、主な原因は肝炎ウイルスの持続感染です。B型(HBV)やC型肝炎ウイルス(HCV)の感染に伴う慢性肝炎から、肝硬変を経て、高い確率で肝臓がんを発症します。治療法にはさまざまな方法がありますが、その効果は十分ではなく、ゲノム情報に基づく発がん分子メカニズムの解明と新たな治療法や予防法の開発が求められています。
今回、共同研究グループは、日本人300例の肝臓がんの腫瘍と正常DNAの全ゲノムの塩基配列情報を次世代シーケンサー(NGS)[3]と東京大学医科学研究所附属ヒトゲノム解析センターのスーパーコンピュータ「SHIROKANE」で解読し、がん細胞のゲノム変異を網羅的に解析しました。データ総量は、約70兆個もの塩基配列情報に上りました。その結果、ゲノム異常は1つの腫瘍あたり平均で約10,000カ所でした。既知のがん関連遺伝子(p16、APC、TERT、CCND1、RB1など)のゲノム構造異常[4]に加えて、新規のがん遺伝子(ASH1L、NCOR1、MACROD2、TTC28など)のゲノム構造異常、HBVとアデノ随伴ウイルス(AAV)[5]の肝臓がんゲノムへの組み込み[6]、遺伝子発現に影響を及ぼす可能性のある非コード領域や非コードRNA[7](NEAT1、MALAT1)の変異も多数検出しました。また臨床背景と相関する新たな変異的特徴(シグネチャー)も同定しました。これらは、肝臓がんの発生や進行に深く関与すると考えられます。また、これらのゲノム情報によって肝臓がんは6つに大きく分類され、肝臓がん術後生存率はこの分子分類によって異なることが分かりました。
本成果は今後、がんのゲノム配列情報に基づいた肝臓がん治療の個別化や新規の治療法・予防法開発へ発展する可能性があります。
成果は、国際科学雑誌『Nature Genetics』(4月11日付け:日本時間4月12日)に掲載されます。
※共同研究グループ
理化学研究所 統合生命医科学研究センター
ゲノムシーケンス解析研究チーム
チームリーダー 中川 英刀(なかがわ ひでわき)
副チームリーダー 藤本 明洋(ふじもと あきひろ)
研究員 古田 繭子(ふるた まゆこ)
医科学数理研究グループ
グループディレクター 角田 達彦(つのだ たつひこ)
国立がん研究センター研究所
がんゲノミクス研究分野
分野長 柴田 龍弘(しばた たつひろ)
ユニット長 十時 泰(ととき やすし)
バイオインフォマティクス部門
部門長 加藤 護(かとうまもる)
東京大学 医科学研究所附属 ヒトゲノム解析センター
教授 宮野 悟(みやの さとる)
東京大学 先端科学技術研究センター
教授 油谷 浩幸(あぶらたに ひろゆき)
広島大学 大学院医歯薬保健学研究院 応用生命科学部門 消化器・代謝内科学
教授 茶山 一彰(ちゃやま かずあき)
和歌山県立医科大学 第2外科
教授 山上 裕機(やまうえ ひろき)
大阪府立成人病センター 消化器外科
名誉院長 石川 治(いしかわ おさむ)
東京女子医科大学 消化器病センター 消化器外科
主任教授 山本 雅一(やまもと まさかず)
北海道大学大学院医学研究科 消化器外科学分野Ⅱ
教授 平野 聡(ひらの さとし)
背景
肝臓がんは、日本における部位別がん死亡者数で、男性では3位、女性では6位です。年間約4万人が肝臓がんと診断され、3万人以上が亡くなっています注1)。特にアジア地域とアフリカ地域で発症頻度が高く、世界全体の部位別がん死亡率では第2位に挙げられています注2)。主な原因は、肝炎ウイルスの持続感染であり、慢性肝炎発症から肝硬変を経ると、高い確率で肝臓がんを発症します。発症要因は、B型肝炎ウイルス(HBV)またはC型肝炎ウイルス(HCV)の感染、アルコール摂取などがありますが、その頻度は世界各地域で異なっています。アジア・アフリカでは、HBV感染による肝臓がんが約70%を占めていますが、日本の肝臓がんの主要因はHCV感染が約60%を占めています。一方、欧米では、HCV感染に加えて、アルコール性肝障害や肥満でみられる脂肪性肝障害からの肝臓がんも多い傾向にあります。
近年のDNA解読技術の飛躍的な進歩に伴い、次世代シーケンサー(Next generation sequencer: NGS)を用いて、さまざまな病気のゲノム変異を包括的に解析することが可能になってきています。がんはゲノム異常が蓄積することによって発症し進行する“ゲノムの病気”であり、世界中でがんの網羅的ゲノム解析やゲノム情報に基づく薬の開発・個別化医療が精力的に行われています。2008年、がんのゲノム変異の全容解明とカタログ化を目指し、世界最大規模のがんゲノム国際共同体である「国際がんゲノムコンソーシアム(ICGC)」が発足しました。共同研究グループもICGCに参加し、肝炎ウイルス関連の肝臓がんのゲノム解析を担当し、研究を進めてきました注3)。ICGCや世界のがん研究機関、がん専門病院では、NGSと情報解析技術を利用した全ゲノムシーケンス解析が精力的に行われています。今後、全ゲノムシーケンスが研究の分野のみならず、診断や個別化医療の分野においても、重要な解析手法になると予測されています注4)。
注1)国立がん研究センターがん対策情報センター「日本のがん最新統計まとめ2013」より
注2)「WHO IARC Estimated cancer incident, mortality and prevalence worldwide 2012」より
注3)2012年5月28日プレスリリース「肝臓がん27例の全ゲノムを解読」
注4)2015年12月9日プレスリリース「がんの全ゲノムシーケンス解析の新たなガイドラインを作成」
研究手法と成果
共同研究グループは、主にウイルス肝炎を背景として発症した日本人300例の肝臓がん組織と同一患者由来の血液からDNAとRNAを抽出し、それらの全ゲノム情報を最新のNGSと東京大学ヒトゲノム解析センターの生命科学専用スーパーコンピュータ「SHIROKANE」を利用して解読し、肝臓がんに起きているゲノム変異を全て同定しました。この研究は、単独のがん種の全ゲノムシーケンス解析数としては世界最大規模となります。解析した塩基配列数は全部で約70兆塩基、情報量としては約300テラバイト(1テラバイトは1兆バイト)以上に上ります。
全ゲノムシーケンス解析の結果、1つの腫瘍あたりのゲノム異常は、平均で約10,000カ所であり、塩基が1つだけ変わる「点突然変異」以外に、さまざまな形態の変異を検出することができました。特に、既知のがん関連遺伝子(p16、APC、TERT、CCND1、RB1など)に加えて、新規のがん関連遺伝子(ASH1L、NCOR1、MACROD2、TTC28など)の染色体構造異常や点突然変異を多数発見し(図1)、前者は周辺の遺伝子の発現を大きく変化させていました。
また、HBVとアデノ随伴ウイルス(AAV)ゲノムの肝臓がんゲノムへの組み込みも検出し、これらのウイルスゲノム挿入によって組み込み部位周辺の遺伝子の発現が変化していることを明らかにしました。さらに、遺伝子発現や構造に影響を及ぼす可能性のある非コード領域の変異(プロモーター[8]変異、転写因子CTCF結合領域の変異など)や非コードRNA(NEAT1、MALAT1)の変異を多数検出しました(図2)。特に、染色体の先端構造を維持して細胞の不死化や染色体の安定性に関わる酵素をコードしている「TERT遺伝子」においては(図1)、ゲノム構造異常、ウイルスゲノムの組み込み、プロモーター領域(非コード領域)の点突然変異、コピー数異常といったさまざまなタイプのゲノム変異が集積しており、肝臓がんにおけるTERT遺伝子の発現を恒常的に上昇させていました。
このようなゲノム異常は、全ゲノムシーケンス解析を多数のがんサンプルについて行わなければ発見できない知見であり、肝臓がんの発症や進行に深く関与するものと考えられます。最後に、これら多数のゲノム変異情報を用いると、肝臓がんは大きく6つに分類され、がん抑制遺伝子TP53に関わる遺伝子群に変異がある肝臓がんは予後が不良である等、肝臓がん予後の生存率がこれらの分類によって大きく異なっていることを明らかにしました。
今後の期待
本成果から、がんのゲノム配列情報に基づいた肝臓がん治療の個別化へ発展する可能性があります。さらに、これらゲノム変異を標的とした肝臓がんの新しい治療法や診断法、予防法の開発にも貢献するものと期待できます。また日本発の本研究は、世界最大規模のがんの全ゲノムシークエンス解析であり、本研究で培われたゲノム解析手法や経験は、がんを始めとするさまざまな疾患の全ゲノムシークエンス解析に応用でき、ゲノム研究とその先にあるゲノム医療の構築に貢献するものと期待されます。
現在、ICGC/TCGA[9]において、世界中のさまざまながんの全ゲノムシーケンスデータを共有して解析する、大規模ながん種横断的研究プロジェクトPCAWG[10]が進行中です。その中でも、今回の日本のチームからの肝臓がんのデータおよび解析と解析インフラの貢献度は、10%以上となっています(全2800例中300例)。今後、PCAWGにおけるがん横断的全ゲノム研究の成果についても期待されます。
原論文情報
- Akihiro Fujimoto, Mayuko Furuta, Yasushi Totoki, Tatsuhiko Tsunoda, Mamoru Kato, Yuichi Shiraishi, Hiroko Tanaka, Hiroaki Taniguchi, Yoshiiku Kawakami, Masaki Ueno, Kunihito Gotoh, Shun-ichi Ariizumi, Christopher P Wardell, Shinya Hayami, Toru Nakamura, Hiroshi Aikata, Koji Arihiro, Keith A Boroevich, Tetsuo Abe, Kaoru Nakano, Kazuhiro Maejima, Aya Sasaki-Oku, Ayako Ohsawa, Tetsuo Shibuya, Hiromi Nakamura, Natsuko Hama, Fumie Hosoda, Yasuhito Arai, Shoko Ohashi, Tomoko Urushidate, Genta Nagae, Shogo Yamamoto, Hiroki Ueda, Kenji Tatsuno, Hidenori Ojima, Nobuyuki Hiraoka, Takuji Okusaka, Michiaki Kubo, Shigeru Marubashi, Terumasa Yamada, Satoshi Hirano, Masakazu Yamamoto, Hideki Ohdan, Kazuaki Shimada, Osamu Ishikawa, Hiroki Yamaue, Kazuaki Chayama, Satoru Miyano, Hiroyuki Aburatani, Tatsuhiro Shibata, and Hidewaki Nakagawa, "Whole genome mutational landscape and characterization of non-coding and structural mutations in liver cancer", Nature Genetics, doi: 10.1038/ng.3547
発表者
理化学研究所
統合生命医科学研究センター ゲノムシーケンス解析研究チーム
チームリーダー 中川 英刀(なかがわ ひでわき)
国立がん研究センター がんゲノミクス研究分野
分野長 柴田 龍弘(しばた たつひろ)
(東京大学医科学研究所 ヒトゲノム解析センター ゲノム医科学分野 教授)
東京大学 医科学研究所附属 ヒトゲノム解析センター
教授 宮野 悟(みやの さとる)
広島大学 大学院医歯薬保健学研究院
応用生命科学部門 医学分野 消化器・代謝内科学
教授 茶山 一彰(ちゃやま かずあき)
報道担当
理化学研究所 広報室 報道担当
Tel: 048-467-9272 / Fax: 048-462-4715
国立がん研究センター 企画戦略局 広報企画室
Tel: 03-3542-2511(代表) / Fax: 03-3542-2545
ncc-admin [at] ncc.go.jp(※[at]は@に置き換えてください。)
東京大学 医科学研究所 管理課総務チーム
Tel: 03-5449-5601
t-soumu [at] ims.u-tokyo.ac.jp(※[at]は@に置き換えてください。)
広島大学 社会産学連携室広報部広報グループ
Tel: 082-424-6762
koho [at] office.hiroshima-u.ac.jp(※[at]は@に置き換えてください。)
補足説明
- 1.全ゲノムシーケンス解析
次世代シーケンサーを使って、個人(約30億塩基)やがんの全ゲノム情報を解読し、配列の違いや変化を同定すること。データが大量になるため、「SHIROKANE」のようなスーパーコンピュータを使って情報解析を行うのが一般的である。全ゲノムシーケンス解析の場合、タンパク質をコードする1~2%の範囲のエクソンだけでなく、遺伝子の発現を制御するゲノム領域の変異やさまざまな構造異常(大きなゲノム配列異常)も検出可能で、究極のゲノム解析手法といえる。がんの場合は、がんのDNAと同一患者由来の正常DNAの全ゲノムシーケンス解析を行い、その差分を調べる。 - 2.国際がんゲノムコンソーシアム(ICGC)
ICGCはInternational Cancer Genome Consortiumの略。がんのゲノム異常の包括的なカタログを作成するという目的を達成するため、2008年に発足した国際連携研究組織。ICGCの各メンバーは、データ収集・解析に関するICGCの共通基準のもと、1種類のがんについて500症例を解析し、ICGCのデータベースに登録して世界中に公開する。2015年時点で、米国の大規模がんゲノムプロジェクト(TCGA)に加えて、ヨーロッパ、南北アメリカ、アジア、オーストラリアの16カ国およびEUの機関や組織が参画し、全体で500億円以上の資金供与の約束がなされ、78種のがんについての大規模ゲノム研究プロジェクトが進められている。これまで、17,867例ものがんのゲノム情報がICGCのポータルサイトで公開され、世界中のがん研究に活用されている。日本からは、理化学研究所と国立がん研究センターが中心となって参画している。
ICGCホームページ(英語) - 3.次世代シーケンサー(NGS)
ヒトゲノムの全配列約30億塩基を1,000米ドル以下のコストで解読すべく、欧米の政府や企業が技術開発を行った結果、より高速高精度の性能を持つシーケンサーが開発された。従来の方法に比べ、超大量のDNAシーケンス反応を並列して行うことができる。今回は主に、6日間で約1兆個(ヒトゲノム10人分)の塩基配列を解読できるスペックのNGSを使用した。 - 4.ゲノム構造異常
1塩基の配列が変化する点突然変異と異なり、数百~数百万塩基の配列が欠失、組み込み、重複、逆位(方向が逆になる)、染色体間で転座する(移動する)など、大きなゲノム配列の変化をいう。全ゲノムシーケンス解析にて、網羅的に検出できるようになった。 - 5.アデノ随伴ウイルス(AAV)
DNAゲノムを保有するウイルスで、ヒトに感染しても重篤な症状を引き起こさず、19番染色体の特定のゲノム領域などに組み込まれることが知られている。AAVを改変したウイルスベクターが、遺伝子治療で使用されている。 - 6.ウイルスゲノムの組み込み
ウイルスの配列がヒトのゲノム配列の入り込む現象のことを指す。HBVの場合、まずウイルスに感染した正常肝臓組織においてHBVゲノム組み込みが起こり、がん化に伴いがん細胞にも引き継がれると考えられている。今回、アデノ随伴ウイルス(AAV)のゲノム配列も数例の肝臓がんで検出され、AAVゲノムの一部ががんゲノムに組み込まれていた。これらのウイルスゲノム配列が TERT遺伝子や MLL4遺伝子などの特定のヒトのゲノム部位に入り込むことも、肝臓がんのがん化に関わっているものと考えられる。 - 7.非コード領域、非コードRNA
ヒトゲノム(約30億塩基)のうち、タンパク質をコードしている遺伝子領域はわずか1~2%であり、残りの領域は非コード領域と呼ばれる。非コード領域には、遺伝子の発現を制御している領域やタンパク質に翻訳されないRNA(非コードRNA)をコードしている領域が含まれ、遺伝子の転写調節やゲノムの複雑な構造を調節しているものと考えられている。 - 8.プロモーター
遺伝子の転写開始付近に位置して、遺伝子を発現させる機能を持つ塩基配列。プロモーターがないと遺伝子は発現しない。 - 9.TCGA(がんゲノムアトラス計画)
TCGAはThe Cancer Genome Atlasの略で、米国主導の大規模ながんゲノム解析プロジェクトである。ゲノム、トランスクリプトーム、エピジェネティックな修飾など、がんのゲノムについて包括的な理解を目指し解析を進めている。 - 10.PCAWG
ICGC/TCGA内のがんの全ゲノムシーケンス解析のデータを集積し、ICGC・TCGAの共同作業にてがんの横断的(PanCancer)解析を行うプロジェクトとして2014年に始動した。最大3000例のさまざまながんの全ゲノムシーケンスのビックデータを東京大学医科学研究所ヒトゲノム解析センターのスパコン「SHIROKANE」を含む10か所のデータセンターで仮想空間を作り、分担して解析を行っている。生データだけで約2ペタバイト(1ペタバイトは1000兆バイト)の情報量になる。PCAWGは、PanCancer Analysis of Whole Genomesの略。
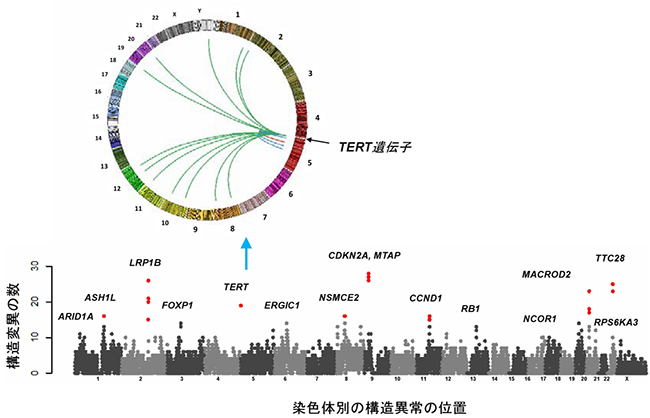
図1 肝臓がんのゲノム構造異常に関わる遺伝子
X軸:染色体(1番~22番、23番:X染色体)ごとの構造異常の位置。
Y軸:検出されたそれぞれの遺伝子に関わる構造異常の数
5番染色体にあるTERT遺伝子においては、さまざまな染色体の部位への転座(遺伝子の一部が本来の位置から移動すること)に加えて、HBVゲノムの組み込みも多数観察され、それらによって、TERT遺伝子の発現が上昇していた。左上の円は、TERT遺伝子の転座部位を示す。赤点は、3%以上の肝臓がんで検出された変異を示している。
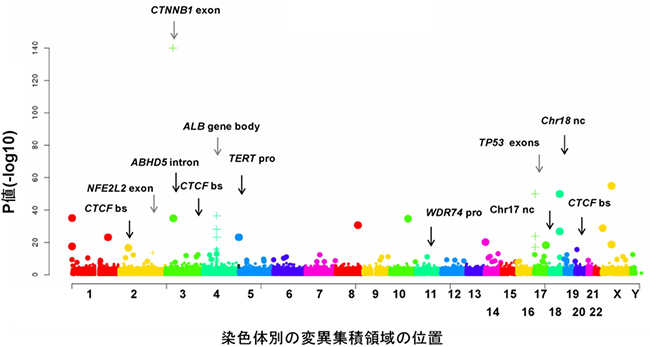
図2 肝臓がんの非コード領域のゲノム変異
X軸:染色体(1番~22番、X染色体、Y染色体)ごとの変異集積の位置。
Y軸:-log10(P値:偶然にそのようなことが起こる確率)で、変異集積の統計学的確からしさの指標。
これまで判明していたCTNNB1遺伝子(β-Catenin)やTP53遺伝子のエクソン(exon)に加えて、多くの非コード領域の特定部位(プロモーターやエンハンサー領域の変異やCTCF結合部位など)に変異が集積していた。CTCF bsは、CTCF因子が結合する部位でゲノムの3次構造を変化させている。TERT proは、TERT遺伝子のプロモーター領域。WDR74proは、WDR74遺伝子のプロモーター領域。Chr17 nc、Chr18 ncは意義不明だが、明らかに変異が集積していた非コード領域。