Apr. 18, 2020
New dataset for drug design uses quantum mechanics-based interactions of new coronavirus proteins and drug candidates
On April 17, a research group led by Teruki Honma of the RIKEN Center for Biosystems Dynamics Research (BDR) and Associate Professor Kaori Fukuzawa of Hoshi University published a dataset on the interactions between SARS-CoV-2—the virus that causes COVID-19—proteins and candidate drug compounds. The data was published as part of the FMO database, a collection of data based on the fragment molecular orbital (FMO) method.
The genome of SARS-CoV-2, like those of other viruses, encodes proteins that form the virion—the virus capsule that allows the virus to infect new cells—as well as enzymes involved in the replication of the virus’s genome and the synthesis of its proteins. Some of the target proteins for potential therapeutic agents include the spike proteins that form virions, nucleocapsid proteins, the main protease—an enzyme that acts inside cells after infecting them, and RNA dependent RNA polymerase.
One important dataset for the development of drugs to target these proteins is a special page of the Protein Data Bank Japan (PDBj), which includes 131 crystal structures as of April 15 for nine of the roughly 20 viral proteins of SARS-CoV-2, either of the isolated proteins or as complexes with drug candidate compounds or human-derived proteins. The structural information is useful, but as a further step it is crucial to make precise calculations in order to help understand how drug candidate molecules bind with the viral proteins.
To make this possible, the research team took the crystal structures and used the FMO method to calculate interactions between various molecules using ab initio quantum mechanical calculations. The FMO method, which was developed in 1999 by Kazuo Kitaura and co-workers, enables researchers to make ab initio quantum mechanical calculations for whole protein molecules by dividing them into fragments such as amino acid residues and drugs. One advantage of the FMO method is that the inter-fragment interaction energy (IFIE) between an amino acid residue and a drug molecule can be calculated and classified into four different types of energy component (PIEDA), including electrostatic and dispersive energies.
They carried out FMO calculations on 65 of the 131 structures registered in PDBj and analyzed their interactions with several drug candidate compounds (various attractive and repulsive forces between the molecules). To do the analysis, the group used RIKEN’s HOKUSAI supercomputer, the Tokyo Institute of Technology’s TSUBAME3.0, and Oakforest-PACS Supercomputer System at the University of Tokyo, using the ABINIT-MP quantum chemistry calculation software, a program developed exclusively for FMO calculations. Tatsuya Nakano of the National Institute of Health Sciences started the development at the beginning of the theoretical development of FMO theory in 1999, and a research group led by Yuji Mochizuki of Rikkyo University has continued the development.
The group has published the data as part of FMODB, the world’s first database of quantum mechanical data for proteins, allowing users to precisely evaluate the mechanism by which a therapeutic drug candidate compound binds to a target protein, and the effect of stronger binding to the target. The database was developed by Honma and Fukuzawa with the FMO drug design consortium and was published in March 2019. (link to FMO Database webpage).
According to Honma, “We are very happy to have been able to publish this data, and hope it will be useful for researchers trying to find therapies to combat this terrible disease.”
The work was performed as part of the special COVID-19 research project organized by RIKEN's Drug Discovery and Medical Platforms Program.
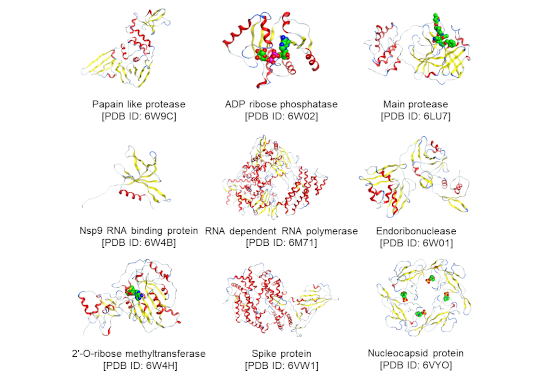
Figure 1. Representative structure of the nine proteins analyzed using FMO method
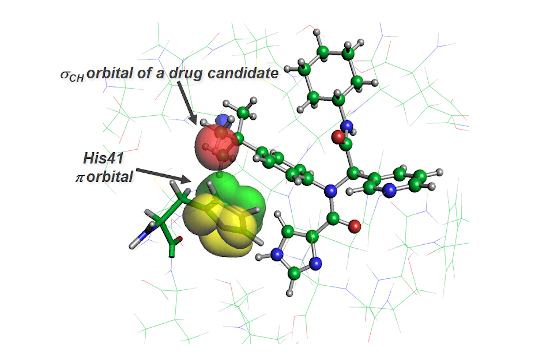
Figure 2. CH/pi orbital interaction between the main protease and a drug candidate
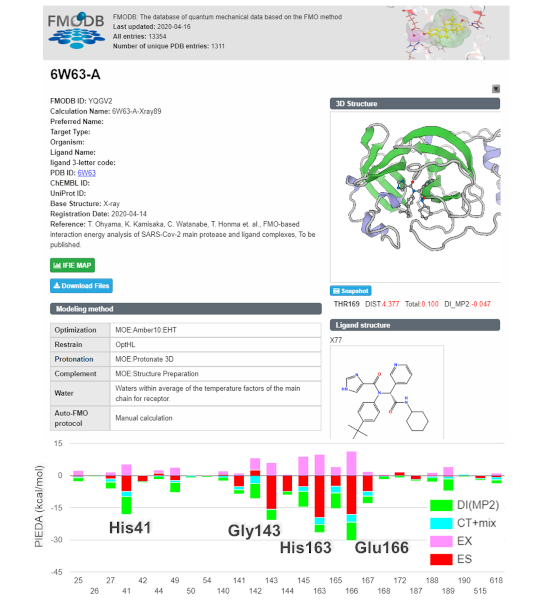
Figure 3. Example of the FMODB data on the interactions between the main protease and a drug candidate compound.