Jan. 10, 2017 Press Release Biology
Blocking obesity with a protein-sugar combination
Researchers at the RIKEN-Max Planck Joint Research Center for Systems Chemical Biology in Japan have discovered an enzyme that can prevent obesity in mice. Published in Journal of Biological Chemistry, the study shows that the enzyme ST6GAL1 prevents the differentiation of fat cells by assisting in the transfer of a key sugar to a protein involved in the differentiation of fat cells.
The function of most proteins can be modified through the attachment or removal of sugar chains called glycans—a process called glycosylation. Because this process affects the development of several diseases that are related to obesity—such as diabetes, emphysema, and cardiovascular disease—a team of glycobiologists led by Naoyuki Taniguchi at the RIKEN-Max Planck Joint Research Center in Japan hypothesized that obesity itself might be linked directly to irregularities in normal glycosylation. To test this theory, they used a mouse model of obesity generated with a high fat diet and looked for changes in the expression levels of glycosyltransferases—the enzymes that make transferring sugars onto proteins possible.
Explains Taniguchi, “By using an unbiased analysis of almost all glycan-synthesizing enzymes, we were able to identify a specific glycan that is altered dramatically during obesity.”
The team examined obese mice and normal mice, and compared gene expression within the fatty tissue of internal organs. They found that levels of the enzyme ST6GAL1 were more than 96% lower in obese mice than in normal mice. Having identified the gene expression that was altered in obesity, the next step was to figure out what exact process was disrupted by the missing ST6GAL1, and that required knowing the target of ST6GAL1’s activity.
Because ST6GAL1 is known to transfer the sugar sialic acid to proteins, they searched for proteins in the obese mice that were modified by sialic acid much less than they were in normal mice. They found one such protein called integrin-β1, which is already known to be involved in the maturation of fat cells.
The team then studied the process in cultures of cells that were induced to become fat cells. They found that St6gal1 expression was indeed reduced during fat-cell differentiation, and that there reverse—preventing fat-cell differentiation—could be achieved by overexpressing St6gal1, which then activated an integrin-β1 pathway.
The team again examined the role of ST6GAL1 in preventing weight gain in vivo. They fed normal mice and St6gal1 knockout nice the same high fat diet, and found that the knockout mice gained much more weight, especially in the fat tissue of their internal organs.
Taniguchi notes that, “the knockout mouse data revealed that ST6GAL1 suppresses adipogenesis in vivo, which will help promote the importance of looking at glycans as regulatory factors for obesity.”
ST6GAL1 likely modifies other proteins besides integrin-β1, and the way it prevents differentiation of fat cells—called adipogenesis—is still unclear.
“Answering these questions is an interesting challenge for the next phase of our research,” says Taniguchi. “In the short-term, we hope to develop a means to upregulate St6gal1 in vivo to prevent obesity in mice. In the longer term, this could lead to new glycan-targeted strategies to cure obesity-related diseases.”
Reference
- Tomoko Kaburagi, Yasuhiko Kizuka, Shinobu Kitazume, and Naoyuki Taniguchi, "Inhibitory Role of α2,6-Sialylation in Adipogenesis", The Journal of Biological Chemistry, doi: 10.1074/jbc.M116.747667
Contact
Team Leader
Naoyuki Taniguchi
Disease Glycomics Team
Systems Glycobiology Research Group
RIKEN-Max Planck Joint Research Center for Systems Chemical Biology
Global Research Cluster
Adam Phillips
RIKEN International Affairs Division
Tel: +81-(0)48-462-1225 / Fax: +81-(0)48-463-3687
Email: pr[at]riken.jp
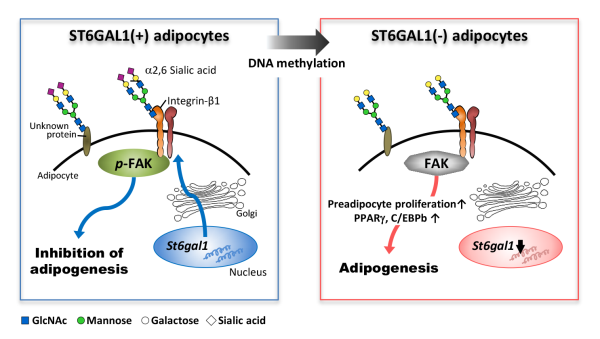
Schematic model for the inhibitory role of ST6GAL1 in adipogenesis