Apr. 30, 2009 Research Highlight Biology
Filling out the map
Recent findings from the FANTOM consortium spotlight new mysteries and challenge old assumptions about the mammalian genome
 Figure 1: The FANTOM4 team—represented in part here—is comprised of scientists at more than 50 research institutions in 15 nations.
Figure 1: The FANTOM4 team—represented in part here—is comprised of scientists at more than 50 research institutions in 15 nations.
Even with a complete sequence at their disposal, scientists are still laboring to unlock the secrets of the human genome—but data from a landmark international research effort headed by the RIKEN Omics Science Center (OSC) in Yokohama offer surprising new insights and promising foundations for future work.
A primary mission of the Functional Annotation of the Mammalian Genome (FANTOM) Consortium (Fig. 1) is to exhaustively catalogue human and mouse genes and their activity. In previous projects, FANTOM has obtained full-length sequence data for over 100,000 mouse gene transcripts, but their latest iteration, FANTOM4, is even more ambitious. “FANTOM4’s main targets have been to demonstrate that it is possible to use sequencing to detect not only DNA or RNA sequences, but also to detect expression and—much more importantly—the networks that control transcription,” explains OSC researcher Harukazu Suzuki.
A key weapon in FANTOM’s arsenal is their ‘deepCAGE’ strategy, combining a method called 5’ cap analysis of gene expression (CAGE), which allows scientists to accurately identify and quantify activity of transcriptional start sites (TSSs), with next-generation sequencing technology. Now, in three new articles from Nature Genetics, FANTOM describes striking findings achieved through their pairing of sophisticated experimental and analytical techniques.
Network news
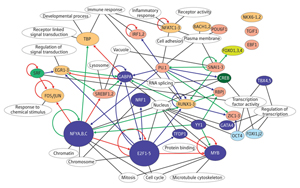 Figure 2: The complicated transcription network uncovered by the latest FANTOM study. Reproduced from Ref. 1 © 2009 Nature Publishing Group
Figure 2: The complicated transcription network uncovered by the latest FANTOM study. Reproduced from Ref. 1 © 2009 Nature Publishing Group
Every gene is regulated by a stretch of DNA called the promoter, containing binding sites for various transcription factor proteins that contribute to gene activation or repression, and whose combined activity ensures that transcription occurs at the right time and place.
The ability to accurately map which factors control which genes and how these regulatory networks interact would be immensely useful in helping scientists to understand the mechanisms underlying virtually any cellular process, and FANTOM achieved major progress on this front with a pilot study investigating chemically induced differentiation of human leukemia cells into mature immune cells1.
The team located promoters throughout the genome based on the clustering of TSSs identified via deepCAGE, and then identified known transcription factor binding sites within those promoters. They collected data from numerous time-points to chart changes in activity during differentiation, and correlated those changes with involvement of specific transcription factors. The result was a detailed, experimentally testable network of regulatory pathways involved in the differentiation process (Fig. 2). “We showed that a network inferred using only experimental data but no previous knowledge can identify all known key transcription factors for THP-1 differentiation and many known—as well as previously unknown—regulatory processes,” says OSC researcher Carsten Daub. “This method can now be applied to biological systems that are poorly understood.”
Double agents
As much as half of the genome is composed of repetitive sequences, derived largely from retrotransposons—DNA elements that can self-replicate and insert themselves into other chromosomal sites with potentially damaging consequences. Scientists have generally looked on these disruptive ‘jumping genes’ uncharitably. “Retrotransposons have been thought mainly to have a parasitic role in the genome,” explains Piero Carninci.
As such, Carninci and his FANTOM4 colleagues were surprised to uncover compelling evidence that the influence of retrotransposons may be far more pervasive—and beneficial—than was previously understood2. Although retrotransposons contain promoters, these were generally assumed to be non-functional and to have little direct effect on gene regulation. However, deepCAGE data revealed that up to 18.1 and 31.4% of TSSs in mice and humans respectively lie within these repetitive elements, and in many cases the positioning of these TSSs suggests that retrotransposon promoters directly contribute to regulation of protein-coding genes.
In addition, more than a quarter of the protein-coding mRNAs examined contained retrotransposon-derived repetitive elements; the presence of these sequences is strongly associated with downregulation of expression, suggesting that retrotransposons may also help ‘silence’ specific genes. Carninci describes these findings as a paradigm shift: “Although the general idea is that retrotransposons are passive—or even harmful— elements of the genome, cells have learned how to use them in symbiotic mechanisms.”
Teeming with tiny transcripts
Little RNAs are big news these days, as scientists continue to uncover an increasingly large array of species of small, non-protein-coding RNA molecules that execute some of the cell’s most important regulatory functions—and FANTOM has now added one more to the list: the tiny transcription initiation RNA (tiRNA)3.
Working in collaboration with University of Queensland investigator John Mattick, a member of the FANTOM consortium, the researchers discovered numerous tiRNAs, averaging 18 nucleotides in length, apparently produced from sites directly adjacent to TSSs in humans, mice, chickens, and even fruit flies. In this last species, up to half of the genes analyzed had tiRNAs associated with them, indicating the ubiquity of these molecules. Subsequent analysis indicated that tiRNAs appear to be deliberately processed by the cell, but not by the same mechanisms used by other known small RNAs classes.
At present it remains unclear exactly what role these molecules fulfill, and further study will clearly be needed. “We do not really know the mechanisms by which they are produced,” says Carninci. “They could be produced by RNA polymerase stalling over promoters, and may be there to influence transcription at this level.”
Outside the CAGE
These findings offer numerous starting points for further studies, and the FANTOM team is following up on the investigation of these phenomena both internally and in collaboration with unaffiliated institutions and researchers. According to Suzuki, RIKEN’s Life Science Accelerator initiative for high-throughput biological analysis will be a direct beneficiary of this work. “We have established a pipeline to analyze transcriptional regulation networks solely by using experimental data without advance knowledge,” he says, “and this pipeline is applicable to any biological event.”
References
- 1. FANTOM Consortium & RIKEN Omics Science Center. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nature Genetics advance online publication 19 April 2009. doi: 10.1038/ng.375
- 2. Faulkner, G.J., Kimura, Y., Daub, C.O., Wani, S., Plessy, C., Irvine, K.M., Schroder, K., Cloonan, N., Steptoe, A.L., Lassmann, T. et al. The regulated retrotransposon transcriptome of mammalian cells. Nature Genetics advance online publication 19 April 2009. doi: 10.1038/ng.368
- 3. Taft, R.J., Glazov, E.A., Cloonan, N., Simons, C., Stephen, S., Faulkner, G.J., Lassmann, T., Forrest, A.R.R., Grimmond, S.M., Schroder, K. et al. Tiny RNAs associated with transcription start sites in animals. Nature Genetics advance online publication 19 April 2009. doi: 10.1038/ng.312
About the Researcher
Carsten Oliver Daub, Harukazu Suzuki and Piero Carninci (from left to right)

Carsten Oliver Daub is the Facility Director of the Life Science Accelerator (LSA) Core Bioinformatics Facility in Omics Science Center. He was born in Berlin, Germany in 1972 and obtained his Chemistry diploma at the Technical University of Berlin and his PhD in Bioinformatics in 2004 at the Max Planck Institute of Molecular Plant Physiology in Potsdam. He has been with RIKEN since April 2006, when he arrived following post-doctoral work in genomics and bioinformatics at the Karolinska Institutet in Stockholm, Sweden. He has led efforts at recruiting bioinformaticians to OSC from across the globe. This strong bioinformatics team has continued to apply distinct and targeted strategies to analyze the transcriptome as they develop new tools to reveal its unknown regulatory functions and readily make these tools available to the scientific community.
Harukazu Suzuki was born in Ono, Fukui prefecture in 1960. His studies were in Pharmaceutical Sciences at Kyoto University where he received his PhD in 1988. He spent a number of years at a major Japanese pharmaceutical company before joining RIKEN’s Genomic Sciences Center in 1998 as a Team Leader. He became a Deputy Project Director in 2005 and Project Director in 2009. He has been a key researcher in the FANTOM consortium since its inception. He guided the FANTOM4 project as scientific coordinator and was one of the papers main authors. His focus is on leading activities for the life sciences analysis pipeline with specific emphasis on modeling of promoter-based expression.
Piero Carninci is the Deputy Project Director of the LSA Technology Development Group and Team Leader of the Functional Genomics Group in Omics Science Center. He was born in Trieste, Italy in 1965 and received his PhD from the University of Trieste in Biological Sciences in 1989. Upon graduation he worked in the National Laboratory of Italian Consortium for Biotechnology and for a private biotechnology venture. He joined the Genome Science Laboratory at RIKEN in April 1995 and since has published prodigiously and received numerous accolades. His work with Dr. Hayashizaki on biotin capping with cap trapper, trehalose applications, and cDNA technology is Nature Milestone 5 for DNA Technologies. In addition to his development of Cap Analysis of Gene Expression (CAGE), which has been integral to the success of the FANTOM projects, he is highly sought after as a world authority on non-coding RNA (see Nature, 19 February 2009) and most recently on repetitive-element associated transcription start sites.