Apr. 2, 2010 Research Highlight Biology
Unearthing the pathways of plasticity
Lipid signaling at the synapse modulates the strength of neuronal communication in the brain
Changes in synaptic strength after repeated communication between neurons is a key mechanism for induction of learning and memory in the brain. Identifying molecules involved in this ‘synaptic plasticity’—and targeting these signaling pathways with drugs—could pave the way to augmenting learning and memory in humans. This is particularly necessary for diseases that have been linked to deficits in synaptic plasticity and memory, such as Alzheimer’s disease. Now, Masao Ito and colleagues at the RIKEN Brain Science Institute in Wako and at the University of Tokyo have shown that lipid signals regulate synaptic plasticity in the cerebellum, a structure at the back of the brain that is involved in motor learning1.
Driving depression
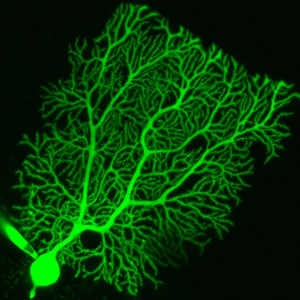 Figure 1: A rat Purkinje neuron injected with a fluorescent dye. Purkinje neurons are the major output neurons of the cerebellum. © 2010 Testuya Tatsukawa
Figure 1: A rat Purkinje neuron injected with a fluorescent dye. Purkinje neurons are the major output neurons of the cerebellum. © 2010 Testuya Tatsukawa
The major output neurons of the cerebellum are called Purkinje neurons (Fig. 1). Repeated electrical stimulation of two separate neuronal inputs onto a Purkinje neuron—called conjunctive stimulation—leads to a reduction in this neuron’s response to subsequent stimulation of one of these inputs. This kind of synaptic plasticity is called long-term depression (LTD) of the neuron’s response. Purkinje neuron LTD has been linked to motor learning, an example of which would be learning to walk and run during early childhood in humans.
During repeated conjunctive stimulation of Purkinje neurons, calcium ions flow into the cell (Fig. 2). These ions activate a cascade of signaling molecules, including phospholipase A2 (PLA2), an enzyme that cleaves lipids in the membrane to release a compound called arachidonic acid (AA). Three subtypes of PLA2 are present in Purkinje neurons.
While two of these isoforms seemed to play no role in LTD, the researchers observed that mice lacking the gene for the third isoform, called cPLA2α, exhibited no LTD in cerebellar brain slices after repeated electrical stimulation. Consistent with this finding, a drug called pyrrolidine-1, which blocks cPLA2α, also inhibited LTD. Because pyrrolidine-1 only inhibited LTD soon after conjunctive stimulation of the Purkinje neurons, the researchers realized that cPLA2α activity was required—but just at that time—for induction of LTD.
Plasticity restored
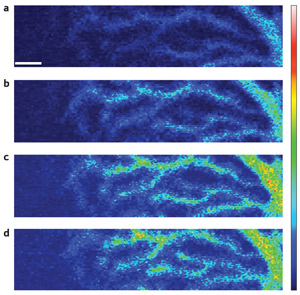 Figure 2: Images showing increasing calcium ion influx into the same area of a Purkinje neuron after conjunctive simulation. Image a was taken 0.5 minutes before simulation, and images b–d were taken at 0.5, 2.5 and 4.5 minutes after the start of simulation, respectively (scale bar, 5 µm). Reproduced from Ref. 1 © 2010 National Academy of Sciences
Figure 2: Images showing increasing calcium ion influx into the same area of a Purkinje neuron after conjunctive simulation. Image a was taken 0.5 minutes before simulation, and images b–d were taken at 0.5, 2.5 and 4.5 minutes after the start of simulation, respectively (scale bar, 5 µm). Reproduced from Ref. 1 © 2010 National Academy of Sciences
Next, Ito and colleagues determined the role of AA in LTD because it is produced by PLA2 along with many other lipid signaling molecules. When they infused AA during conjunctive stimulation of the cerebellar slices, they found it rescued LTD in mice lacking the gene for cPLA2α. However, adding AA onto cerebellar slices that had not been conjunctively stimulated failed to induce LTD on its own. This suggests that LTD also requires other signaling pathways induced by conjunctive stimulation.
Delving deeper into the machinations of the pathway, the researchers then examined the role of cyclooxygenase-2 (COX-2), which is an enzyme that acts on AA to produce additional lipid signaling molecules. They observed that, as with cPLA2α inhibitors, COX-2 inhibitors blocked induction of LTD by repeated conjunctive stimulation of Purkinje neurons. Because the time during which COX-2 inhibitors could block LTD—right after conjunctive stimulation, but not later—was equivalent to the time during which the cPLA2α inhibitors were able to prevent LTD, the researchers conclude that both enzymes, COX-2 and cPLA2α, participate in the same signaling pathway to drive LTD induction.
Investigating further, Ito and colleagues then focused on prostaglandin (PG), which COX-2 produces from AA. They observed that PG rescued LTD not only in the presence of COX-2 inhibitors, but also in brain slices lacking the cPLA2α gene. This suggests that this lipid signaling pathway induces LTD owing to its eventual production of PG. However, when the researchers treated conjunctively stimulated cerebellar slices from normal mice with PG, LTD was not enhanced any further. The team postulates that the PG that is created within the cerebellum during conjunctive stimulation—the ‘endogenous’ PG—induces as much LTD as is possible, and this is why adding extra PG will have no additional effect on further augmenting LTD.
PG binds to and activates various types of prostanoid receptors on the surface of cells. Interestingly, though, when the researchers treated conjunctively stimulated cerebellar slices with presently available inhibitors of these receptors, none of them affected LTD. Ito suspects “that the LTD induction is mediated by an as-yet-unidentified prostanoid receptor or by a direct interaction of PG with receptors for the excitatory neurotransmitter glutamate, which mediate the response of Purkinje neurons to stimulation of one of their inputs.”
The eyes have it
Ito and colleagues next tested whether this lipid signaling pathway, which plays a key role in LTD induction, is also involved in the optokinetic eye movement response (OKR), a type of motor learning of the eye.
When observing an oscillating screen, mice learn to increase their eye movements over time as they follow the motion of the screen. This is called OKR adaptation. Scientists can measure these eye movements and then calculate the rate of OKR adaptation. In mice treated with COX-2 inhibitors, Ito and colleagues observed very low adaptation rates, suggesting that their motor learning of OKR was less robust than in untreated animals. Because COX-2 inhibitors also blocked LTD, these findings provide a strong link between defects in LTD and motor learning dysfunction.
If this signaling pathway is also involved in other types of learning and memory, targeting the pathway with drugs that activate it could be a way to drive learning and memory during disease. “Although it is still unclear how LTD is converted to a permanent memory,” says Ito, “our findings add a novel element to the intricate signal transduction pathways for LTD induction.”
References
- 1. Le T.D., Shirai, Y., Okamoto, T., Tatsukawa, T., Nagao, S., Shimizu, T. & Ito, M. Lipid signaling in cytosolic phospholipase A2α–cyclooxygenase-2 cascade mediates cerebellar long-term depression and motor learning. Proceedings of the National Academy of Sciences USA 107, 3198–3203 (2010). doi: 10.1073/pnas.0915020107
About the Researcher
Masao Ito

Masao Ito was born in Nagoya, Japan, in 1928. He graduated from University of Tokyo with an MD in 1953, and received his PhD in 1959 from the same institution. Until 1962, we worked in the John C. Eccles laboratory at the Australian National University. He then returned to the University of Tokyo as an associate professor, became professor in 1970, and later served as medical dean from 1986 to 1988. He joined RIKEN in 1989, and became the founding director of the Brain Science Institute in 1997. After leaving the post in 2003, he has acted as a senior advisor. His achievements include discovery of the exclusive inhibitory nature of cerebellar Purkinje cells, establishment of the vestibuloocular reflex as a model system of cerebellar motor adaptation, and experimental verification of long-term depression in Purkinje cells and analyses of its signal transduction mechanism.