Apr. 19, 2013 Research Highlight Biology
Understanding metabolism through computer predictions
Combining computer-based statistical analysis with mathematical modeling can improve our understanding of metabolic pathways
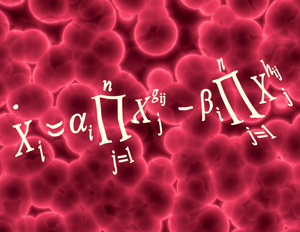 Figure 1: Starting with time-series data of metabolite concentrations, Hirai’s team used a combination of statistical analysis and computer modeling to predict reaction networks for different metabolites within a metabolic system. Reproduced from Ref. 1 © 2013 K. Sriyudthsak et al. (equation); © 2013 Hemera/ Thinkstock (background)
Figure 1: Starting with time-series data of metabolite concentrations, Hirai’s team used a combination of statistical analysis and computer modeling to predict reaction networks for different metabolites within a metabolic system. Reproduced from Ref. 1 © 2013 K. Sriyudthsak et al. (equation); © 2013 Hemera/ Thinkstock (background)
The chemical interactions among molecules and enzymes in an organism’s metabolic system drive growth, reproduction and even evolution. Understanding the pathways that link enzymes and the molecular products of metabolism, called metabolites, can help scientists control metabolic reactions and therefore enhance organism productivity. This can be particularly useful in agriculture and industry.
It is already possible to show whether enzymes are activated or inhibited by certain metabolite interactions through intensive laboratory experiments, and correlations among metabolites can be deduced through statistical analysis of metabolite concentrations in a living organism. However, complete reaction networks of correlated metabolites remain difficult to identify.
Masami Yokota Hirai and Kansuporn Sriyudthsak at the RIKEN Plant Science Center*, in collaboration with Fumihide Shiraishi from Kyushu University in Japan, have now developed a method involving a combination of mathematical modeling and statistical techniques that allows probable metabolic reaction networks to be predicted and identified1.
“In order to gain insight into the molecular mechanisms and biological functions of metabolism,” says Hirai, “we have combined two important approaches to comprehensively understand metabolic reaction networks.”
The team used time-series data of metabolite concentrations to predict the metabolic reaction network for the bacteria Lactococcus lactis. A disturbance in the concentration of one type of metabolite has a knock-on effect on the concentrations of other metabolites in the system. Consequently, analyzing the changes in metabolite concentrations over time can help to produce a picture of the metabolic reaction network.
Firstly, Hirai and her team carried out data smoothing through regression analysis. Next they used a test known as Granger causality to determine if one metabolite’s set of time-series data could be used to forecast that of another metabolite. In this way, the team was able to eliminate metabolites that were unrelated.
The researchers then constructed a mathematical model based on the remaining metabolites to create a probable reaction network. The resulting predictions correlated closely with pathways already established for the bacteria.
“Our novel approach not only predicts metabolic pathways, but the mathematical model can also simultaneously analyze metabolism to determine its characteristics,” explains Hirai. “For example, the model can find metabolic bottlenecks—where a healthy metabolic system is limited in some way—and can also predict the genes or enzymes responsible for causing them.”
The team believes that their combined modeling approach could prove useful in large-scale metabolism studies in the future, and could also help in measuring individual enzyme activities and identifying entirely new metabolic pathways.
* Reorganized into a new center from April 2013
References
- 1. Sriyudthsak, K., Shiraishi, F. & Hirai, M.Y. Identification of a metabolic reaction network from time-series data of metabolite concentrations. PLoS ONE 8, e51212 (2013). doi: 10.1371/journal.pone.0051212