Apr. 18, 2014 Research Highlight Biology
Making sense of the cellular rulebook
Atlases of functionality for specific genomic elements across a diverse range of cell types reveal insights into how different tissues establish their biological and functional identities
 Figure 1: A group photo of scientists from the FANTOM5 consortium at a meeting in the winter of 2011. © 2011 RIKEN Center for Life Science Technologies
Figure 1: A group photo of scientists from the FANTOM5 consortium at a meeting in the winter of 2011. © 2011 RIKEN Center for Life Science Technologies
The cell is an immensely complex biological system involving a multitude of components that work together to drive the cellular machine. Identifying how all of the components fit together in any given cell type is a challenge in itself—integrating the pieces into a functional whole across a wide variety of cell types is an undertaking on a different scale entirely. Yet this is the ambitious goal of the international FANTOM5 consortium, led by Alistair Forrest, Piero Carninci and colleagues from the RIKEN Center for Life Science Technologies and Yoshihide Hayashizaki from the RIKEN Preventive Medicine & Diagnosis Innovation Program, which has made important progress in assembling a functional blueprint for the myriad genomic elements that control gene expression across hundreds of different mammalian cell types1,2.
Nearly 15 years ago, the first iteration of the Functional Annotation of the Mammalian Genome (FANTOM) project set out to identify every gene that undergoes active transcription to produce an RNA message. The recent fourth-generation FANTOM4 consortium combined experimental techniques and computational tools to identify the interactions between various transcription factor proteins and the promoter DNA sequences that regulate gene function. As a proof of concept, the consortium examined how regulatory pathways interact to drive maturation in a class of immune cells known as monocytes. “Through integration of binding site predictions and expression levels of RNA transcripts, we were able to predict key changes in transcription factor activities during differentiation,” says Forrest.
The FANTOM5 consortium (Fig. 1) is expanding this analysis to a far greater scale, encompassing roughly 200 different cell types derived from human and mouse tissue samples. “The aim is to build transcriptional regulatory network models for the majority of mammalian cell types,” says Forrest.
Seeking similarities and differences
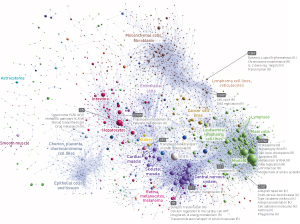 Figure 2: A graphic map of ‘clusters’ of promoters (spheres) that are associated with shared gene expression patterns in various different tissues. Reproduced, with permission, from Ref. 1 © 2014 The FANTOM Consortium and the RIKEN PMI and CLST (DGT)
Figure 2: A graphic map of ‘clusters’ of promoters (spheres) that are associated with shared gene expression patterns in various different tissues. Reproduced, with permission, from Ref. 1 © 2014 The FANTOM Consortium and the RIKEN PMI and CLST (DGT)
A research effort of this magnitude—involving many thousands of samples, prepared and examined by 260 scientists in 20 countries—requires powerful analytical tools. Forrest’s team designed a computational platform called ZENBU to simplify collaborative analysis of such large volumes of experimental data. Although the FANTOM5 project also examined numerous cultured cell lines, the primary focus was on cells isolated from human donor tissue, requiring standardized workflows for analyzing tiny amounts of RNA from small numbers of cells without introducing biases that might skew the data.
The researchers employed a variant of cap analysis of gene expression (CAGE), a technique developed by RIKEN scientists as a means to home in on active genes by sequencing the beginnings of RNA transcripts. Using ZENBU and other tools to map these sequences back to the genome, the researchers identified peaks of activity representing likely transcription start sites (TSSs) for nearly 94% of the known human genes.
Many promoters were associated with multiple TSSs that exhibited different activity levels in different cell types. More generally, the vast majority (80%) of human TSSs showed strong tissue specificity, exhibiting activity in fewer than half of the various cell types profiled (Fig. 2). “Mammalian promoters are often complex entities consisting of tightly packed independent transcription initiation regions with different cell-type-specific preferences,” says Forrest. The researchers were able to identify different combinations of transcription factors that manage this specificity at various promoters.
Promoters are generally close to the TSS, but transcription is also modulated by ‘enhancer’ sequences that can be relatively distant. Interestingly, many enhancer sequences give rise to short RNAs of unknown function, called enhancer RNAs (eRNAs), which made it possible for the FANTOM5 consortium to profile these genomic elements with CAGE. FANTOM5 collaborators Albin Sandelin from the University of Copenhagen in Denmark and Michael Rehli from the University Hospital Regensburg in Germany spearheaded this analysis with Forrest and Carninci. The results correlated closely with other known predictors of enhancer location. As eRNA production appears to be primarily restricted to active enhancers, the FANTOM5 group was able to identify large numbers of tissue-specific enhancers as well as a small but notable subset that acts broadly across cell types.
Part of a bigger picture
These studies represent only the first round of data from the FANTOM5 project, but the clinical possibilities are already tantalizing. For example, preliminary analysis suggests that numerous genetic variations that have been linked to human disease but lie outside of known gene-coding regions may instead affect enhancers characterized by FANTOM5.
“One enhancer variant associated with diabetes led to a 50% reduction in enhancer activity, while another associated with Crohn’s disease led to a 10% reduction in enhancer activity,” says Forrest. Such insights could help scientists assign definite functions to the many enigmatic mutations routinely uncovered in clinical genomics research.
The next stage should yield even greater biomedical dividends. Building on the present work, which focuses on cells ‘at rest’, the consortium is now investigating shifts in gene activity associated with normal biological processes such as growth and development as well as external triggers such as infection. “We are looking at the series of events that happen as a cell transitions from one stable state to another at the level of promoters, enhancers and transcription factors,” says Forrest. A spin-off project from FANTOM5 aims to conduct a similar analysis for cancer in an effort to identify perturbations in gene expression networks that contribute to tumor formation and growth.
More generally, the outputs of the FANTOM5 project will also be used to bolster and extend the utility of data from other large-scale research efforts, such as the detailed genomic map produced by the Encyclopedia of DNA Elements (ENCODE) consortium based in the United States. “This integration is already happening,” says Forrest, “and I think that the FANTOM5 dataset will be used as a reference expression atlas for years to come.”
References
- 1. The FANTOM Consortium and the RIKEN PMI and CLST (DGT). A promoter-level mammalian expression atlas. Nature 507, 462–470 (2014). doi: 10.1038/nature13182
- 2. Andersson, R., Gebhard, C., Miguel-Escalada, I., Hoof, I., Bornholdt, J., Boyd, M., Chen, Y., Zhao, X., Schmidl, C., Suzuki, T. et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461 (2014). doi: 10.1038/nature12787
About the Researcher
Functional Annotation of the Mammalian Genome (FANTOM)

FANTOM is an international research consortium established in 2000 by Yoshihide Hayashizaki and his colleagues to assign functional annotations to the full-length complementary DNAs that were collected during the RIKEN Mouse Encyclopedia Project. FANTOM has since developed and expanded over time to encompass the fields of transcriptome analysis. The project is moving steadily up the layers in the system of life, progressing from an understanding of the ‘elements’—the transcripts—to the ‘system’ of an individual life form—the transcriptional regulatory network. Representing the latest phase of the FANTOM project, FANTOM5 expands on previous efforts, with the aim of generating both a map of the majority of human promoters and comparative transcriptional regulatory network models of each cellular state.