A team from the RIKEN Center for Biosystems Dynamics Research in Osaka, Japan, have used the MDGRAPE-4A dedicated drug discovery supercomputer to analyze the structural dynamics of the main protease of SARS-CoV-2, the virus that causes the new coronavirus disease, COVID-19. The raw data from the simulation has been published on Mendeley Data for use by researchers around the world.
Proteases are one of the main types of enzymes, along with transcriptases and integrases, that viruses use to create new replicas of themselves. The protease allows the virus to reconstitute a new functional virus that can function outside the cell it has infected, by cleaving a large molecule called a polyprotein into individual, functional proteins. These enzymes are targets for protease inhibitors such as saquinavir and ritonavir, which are used to treat HIV infection.
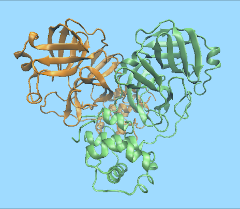
The team used recent x-ray crystallography data on the structure of the new virus’s main protease published by Liu et al (Data on the RCSB Protein Data Bank), to create a molecular dynamics model, composed of 98,694 atoms including 29,712 water molecules. Computing the 10-microsecond-long simulation of the system took ten days on the MD-GRAPE-4A computer. The data show how protease exhibits relaxation and fluctuation in an aqueous solution. The BDR scientists hope that this data will help other researchers in the development of drugs that can target the SARS-CoV-2 virus’s main protease. The data can be accessed from Mendeley Data.