Apr. 22, 2017 Press Release Biology Medicine / Disease
Promising mouse model for a devastating genetic deficiency
Researchers from the Global Research Cluster in Japan have developed a potential mouse model for the genetic disorder known as an NGLY1 deficiency. Published in the journal PLOS Genetics, the study describes how a complete knockout of the Ngly1 gene in mice leads to death just before birth, which can be partially rescued by a second knockout of another gene called Engase. When related genes in the mice used for making the knockouts are variable, the doubled-deletion mice survive and have symptoms that are analogous to humans with NGLY1-deficiency, indicating that these mice could be useful for testing potential therapies.
NGLY1-deficiency is a relatively newly discovered genetic disorder, with the first patient identified in 2012. The symptoms are severe, and include delayed development, disordered movement, low muscle tone and strength, and the inability to produce tears. Understanding how lack of NGLY1 leads to these symptoms is critical when considering targets for therapeutic interventions, and creating useful animal models of the disease is therefore equally important.
The RIKEN team has already had some success (published on PNAS) studying the consequences of Ngly1 deficiency in cultured animal cells. The Ngly1 gene codes for an enzyme that helps remove sugar chains from proteins that are scheduled for degradation. Their research showed that when Ngly1 was absent, sugars normally removed by Ngly1 were improperly removed by another enzyme called ENGase. Knocking out the ENGase gene led to normal protein degradation.
In the current study, the researchers first examined the effects of knocking out Ngly1 in mice. They found that when mice lacked both Ngly1 genes—one from each parent—they always died just before birth. However, a double knockout of both Ngly1 and ENGase genes resulted in mice that survived after birth, but not for very long.
This positive result was actually unexpected. “We thought that ENGase acted further downstream to Ngly1 in the catabolism of glycoproteins” notes team leader Tadashi Suzuki, “and were surprised when the double knockout was able to suppress the lethality of Ngly1-KO mice. If ENGase was merely an enzyme downstream of Ngly1, nothing should have happened. This was truly a case of serendipity.”
Although the double knockout mice survived, they shared several defects that are similar to the symptoms observed in people with NGLY1-deficiency. As these mice aged, they developed characteristics such as bent spines, trembling, limb-clasping and shaking, and by 45 weeks, the survival rate was reduced to 60%. These mice could therefore be useful model mice for developing treatments for the human genetic disorder.
Another factor affecting survival and symptoms turned out the be the genetic background of the mice, that is, the genotypes of all genes related to Ngly1 and Engase, which likely affect how they function. When mice were crossed with an outbred strain, the single Ngly1 knockout proved less lethal, and the double knockout proved to be even more helpful, with mice only showing hind-limb clasping after 30 weeks.
These findings show that the biological processes involved are quite complicated. Despite this however, it is clear that preventing ENGase from acting can alleviate symptoms of Ngly1 deficiency in mice. Figuring out which aspects of the genetic background help reduce symptoms is perhaps a long-term goal.
Although the condition was only recently discovered, research into NGLY1-deficiency has been facilitated through efforts from the Grace Science Foundation, and the RIKEN team has recently begun a collaboration (article on Asian Scientist) with Takeda Pharmaceuticals and T-CiRA at Kyoto University.
“For now, the next step,” says Suzuki, “will be to isolate an in vivo inhibitor for ENGase and determine whether it can improve the symptoms related to NGLY1-deficiency. As we do not know much about the pathophysiology of the disorder, this might help us find potential targets for therapy, but also might lead to a better understanding of other diseases. Such chains of unexpected results are the beauty of basic science!”
Reference
- Haruhiko Fujihira, Yuki Masahara-Negishi, Masaru Tamura, Chengcheng Huang, Yoichiro Harada, Shigeharu Wakana, Daisuke Takakura, Nana Kawasaki, Naoyuki Taniguchi, Gen Kondoh, Tadashi Yamashita, Yoko Funakoshi, and Tadashi Suzuki, "Lethality of mice bearing a knockout of the Ngly1-gene is partially rescued by the additional deletion of the Engase gene", PLOS Genetics, doi: 10.1371/journal.pgen.1006696
Contact
Team Leader
Tadashi Suzuki
Glycometabolome Team
Systems Glycobiology Research Group
Global Research Cluster
Team Leader
Shigeharu Wakana
Technology and Development Team for Mouse Phenotype Analysis
RIKEN BioResource Center
Adam Phillips
RIKEN International Affairs Division
Tel: +81-(0)48-462-1225 / Fax: +81-(0)48-463-3687
Email: pr[at]riken.jp
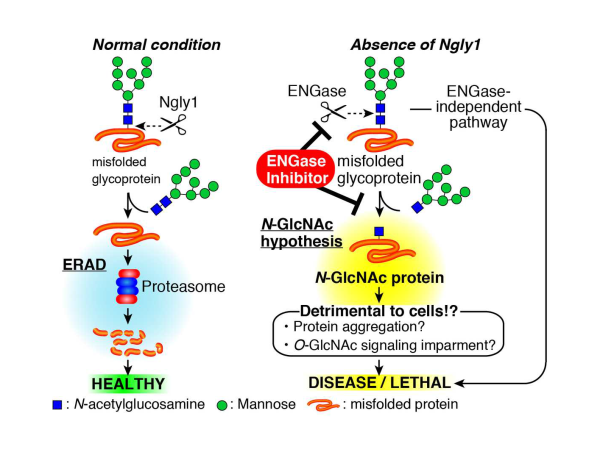
N-GlcNAc hypothesis and the accumulation of N-glycoproteins in the cytoplasm of Ngly1−/−;Engase−/− MEF cells
Schematic representation of an N-GlcNAc hypothesis and therapeutic treatment of NGLY1-deficiency based on ENGase inhibition. In the absence of Ngly1, ENGase acts on some portions of unfolded glycoproteins to form N-GlcNAc proteins. The presence of an excess of N-GlcNAc proteins somehow results in detrimental effects on cells/mice.