May 11, 2018 Research Highlight Biology
New single-cell RNA sequencing methods could lead to better regenerative therapies
An improved method for analyzing the expression of genes by single cells promises to enhance regenerative medicine therapy as well as disease research
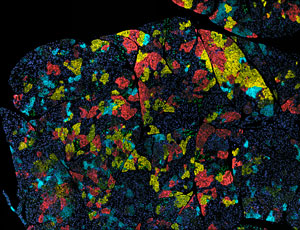 Figure 1: Tumors, such as this one from a mouse model of pancreatic cancer, consist of a mixture of cells with different genetic mutations (indicated by different colors). An improved method for sequencing RNA of single cells will help scientists identify such cellular heterogeneity. © Abramson Cancer Center at the University of Pennsylvania/NATIONAL CANCER INSTITUTE/SCIENCE PHOTO LIBRARY
Figure 1: Tumors, such as this one from a mouse model of pancreatic cancer, consist of a mixture of cells with different genetic mutations (indicated by different colors). An improved method for sequencing RNA of single cells will help scientists identify such cellular heterogeneity. © Abramson Cancer Center at the University of Pennsylvania/NATIONAL CANCER INSTITUTE/SCIENCE PHOTO LIBRARY
An more effective and affordable way of analyzing the expression of genes at the single-cell level will assist scientists and clinicians to find rare cell types within mixed populations of cells such as tumors1.
RNA sequencing of single cells has emerged as an invaluable tool for teasing apart the contributions of different cell types or cell states in complex biological arrangements like those found in tumors, the brain and immune repertoires. This knowledge is vital for discovering how diseases develop.
In 2013, Itoshi Nikaido of the RIKEN Center for Biosystems Dynamics Research and his colleagues developed a method called Quartz-Seq to simplify and improve existing techniques for single-cell RNA sequencing. Now, they have updated the method to make it more effective and affordable. Known as Quartz-Seq2, this new method will be valuable for ensuring the consistency of transplanted cells delivered to patients receiving regenerative medicine therapy. It will also aid research into disease dynamics and progression.
“We hope that Quartz-Seq2 will contribute to the work of the single-cell biology research community and the medical field,” says Nikaido, who led the tool’s development.
Quartz-Seq2 involves a series of steps. A population of cells is sorted and separated into individual cell compartments. All the RNA in the cells is then converted to DNA, and each cell’s DNA is tagged with a unique barcode. That DNA is collected, amplified and sequenced. The unique molecular identifiers assigned to each RNA molecule allow scientists to detect any differences in gene expression that indicate differences between cells.
 Itoshi Nikaido (left), with first co-authors Yohei Sasagawa (center) and Hiroki Danno (right) and other co-workers has developed an improved way of analyzing gene expression at the single-cell level. © 2018 RIKEN
Itoshi Nikaido (left), with first co-authors Yohei Sasagawa (center) and Hiroki Danno (right) and other co-workers has developed an improved way of analyzing gene expression at the single-cell level. © 2018 RIKEN
Nikaido’s team demonstrated the superiority of Quartz-Seq2 over other techniques by analyzing a population of about 9,000 mouse embryonic stem cells cultured in a lab dish. They then characterized around 1,000 cells taken from the fat tissue of a living mouse, and identified two closely related types of stem cells therein, each with distinct characteristics.
These examples show the potential of the method, says Nikaido. More broadly, though, “this heterogeneity information will help us to treat human disorders.”
Nikaido and his collaborators also recently described another single-cell RNA sequencing method, called RamDA-seq2. This method is more expensive and cumbersome than Quartz-Seq2, but it can decode full-length RNA transcripts—something needed for certain types of analysis, such as when researchers are interested in RNA splicing patterns or expression of non-polyA RNAs.
With both Quartz-Seq2 and RamDA-seq, the research community now has a wealth of options for studying RNA variation at the single-cell level. “Both methods show great quantitative performance,” notes Nikaido.
Related contents
- Researchers successfully sequence total RNA of single cells
- Looking at every cell (part 1)
- Perusing the cellular library
- Linking theory and experiment with bioinformatics
References
- 1. Sasagawa, Y., Danno, H., Takada, H., Ebisawa, M., Tanaka, K., Hayashi, T., Kurisaki, A. & Nikaido, I. Quartz-Seq2: a high-throughput single-cell RNA-sequencing method that effectively uses limited sequence reads. Genome Biology 19, 29 (2018). doi: 10.1186/s13059-018-1407-3
- 2. Hayashi, T., Ozaki, H., Sasagawa, Y., Umeda, M., Danno, H. & Nikaido, I. Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs. Nature Communications 9, 619 (2018). doi: 10.1038/s41467-018-02866-0