Nov. 29, 2019 Research Highlight Biology Physics / Astronomy
Faster modeling of interactions between ligands and proteins
An improved simulation technique for modeling how small molecules interact with proteins could help the development of new drugs
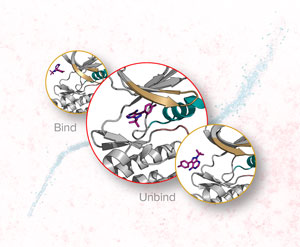 Figure 1: RIKEN researchers have used an enhanced molecular dynamics simulation to simulate the interaction between tyrosine kinase Src, a signaling protein in cells that can turn on protein synthesis and cellular growth, and an inhibitor (magenta), which inhibits the protein by binding to it. Overexpression of the gene that codes for tyrosine kinase Src has been linked with breast, ovarian and colon cancer. The background image shows a colored transmission electron micrograph of tyrosine kinase Src. Credit: Insets: © 2019 RIKEN Center for Biosystems Dynamics Research; background image: © LINDA BARTLETT/NATIONAL CANCER INSTITUTE/SCIENCE PHOTO LIBRARY
Figure 1: RIKEN researchers have used an enhanced molecular dynamics simulation to simulate the interaction between tyrosine kinase Src, a signaling protein in cells that can turn on protein synthesis and cellular growth, and an inhibitor (magenta), which inhibits the protein by binding to it. Overexpression of the gene that codes for tyrosine kinase Src has been linked with breast, ovarian and colon cancer. The background image shows a colored transmission electron micrograph of tyrosine kinase Src. Credit: Insets: © 2019 RIKEN Center for Biosystems Dynamics Research; background image: © LINDA BARTLETT/NATIONAL CANCER INSTITUTE/SCIENCE PHOTO LIBRARYA computational method for simulating the interaction between small molecules and proteins has been enhanced by an all-RIKEN team1. This improvement promises to boost the speed and accuracy of designing new drugs.
Many biological processes are regulated by the interactions between small molecules known as ligands and large proteins. Since many drugs work by mimicking the interactions of natural ligands with proteins, it is vital to accurately simulate such interactions when designing new drugs.
Molecular dynamics simulations have been used extensively to simulate interactions between ligands and proteins. But even with today’s supercomputers, they can only simulate a few binding and unbinding events between a protein and a ligand, whereas in reality a ligand can interact with a protein in various different ways.
Now, Yuji Sugita and Suyong Re and co-workers at the RIKEN Center for Biosystems Dynamics Research have extended a molecular dynamics simulation using the replica-exchange molecular dynamics method to allow it to compute hundreds of binding and unbinding events.
The team demonstrated its potential by simulating the interaction between an inhibitor and a protein kinase—a class of proteins that has a large range of functions and has been implicated in various diseases, including cancer. Their enhanced technique was able to simulate the interaction, which involved about 100 binding and unbinding events. The calculation took about a month using the K computer. In contrast, conventional molecular dynamics simulation techniques would take at least ten times longer.
The method has other advantages besides speed. “We don’t need prior knowledge of the bound-state interactions, which is often brought from well-defined crystal structures obtained using x-ray diffraction, to search for compounds that bind to proteins,” notes Re.
It will also enable researchers to investigate binding that occurs early in the interaction. “Much prior work on drug design has focused on the final bound state, but our work suggests that early bound states are also important since they can change the fate of drugs,” explains Re. “If we can calculate those early bound states, the possibility of drug design could increase significantly—this is a new idea.”
It is an exciting time for the group since RIKEN is due to introduce a new supercomputer, Fugaku, in 2021 to replace the K computer. “We’re expecting Fugaku to be more than 100 times faster than the K computer,” says Sugita. “A target that might take more than a month to simulate using the K computer might take a day with Fugaku.”
The team has ambitious plans. “Our next step is to simulate protein–ligand interactions in a concentrated protein solution,” says Sugita. “But ultimately we’d like to study drug discovery in a living cell—that’s a totally new world.”
Related content
Reference
- 1. Re, S., Oshima, H., Kasahara, K., Kamiya, M. & Sugita, Y. Encounter complexes and hidden poses of kinase-inhibitor binding on the free-energy landscape. Proceedings of the National Academy of Sciences USA 116, 18404–18409 (2019). doi: 10.1073/pnas.1904707116