Jan. 10, 2014 Research Highlight Biology
Finding meaning in gene expression ‘noise’
Analysis of gene expression at the single-cell level reveals patterns and pathways that are impossible to detect in larger-scale experiments
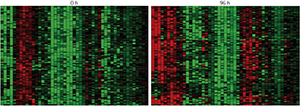 Figure 1: Analysis of changes in the expression of different genes (columns) in individual cells (rows) reveals clear inter-individual differences but also striking patterns associated with the differentiation process. Red and green squares denote higher and lower gene activity, respectively. These two maps were taken at 0 hours (left) and 96 hours (right), showing the different expression patterns at two cellular states. © 2013 Jay Shin, RIKEN Center for Life Science Technologies
Figure 1: Analysis of changes in the expression of different genes (columns) in individual cells (rows) reveals clear inter-individual differences but also striking patterns associated with the differentiation process. Red and green squares denote higher and lower gene activity, respectively. These two maps were taken at 0 hours (left) and 96 hours (right), showing the different expression patterns at two cellular states. © 2013 Jay Shin, RIKEN Center for Life Science Technologies
Static models of gene regulation networks are inevitably oversimplified, indicating how one gene specifically switches certain target genes on while turning others off. However, the reality is much more dynamic and thus noisier. Jay Shin and colleagues from the RIKEN Center for Life Science Technologies have now monitored one such network in individual cells at different time-points during the complex process of cellular maturation1.
The research team completed their study in a leukemia-derived cell line called THP-1 that can be chemically induced to differentiate into mature immune cells known as macrophages. Four years ago, scientists from the RIKEN-initiated FANTOM (Functional Annotation of the Mammalian Genome) consortium mapped out the genes that participate in this process, but the data were collected from large numbers of cells and thus sacrificed valuable information. “When cells are pooled,” explains Shin, “the variation in gene expression is lost and only the mean expression of the population can be measured. By profiling single cells, we can quantify this natural variation.”
Shin’s team collected gene expression data from 120 individual cells at eight different time-points of THP-1 cell differentiation, focusing on a subset of 45 genes previously identified by the FANTOM research. Their analysis revealed how expression shifted for these genes over time (Fig. 1). However, the researchers also saw highly coordinated patterns of ‘noise’ in gene expression from individual cells at each time-point, revealing well-defined differentiation programs that can be initiated when the underlying gene networks are appropriately rewired.
Other patterns that would likely be lost in a bulk analysis also emerged. For example, subtle variations in the expression of ‘master regulators’ at the top of a gene network were found to trickle down to yield far greater noise in genes downstream. Noise analysis also revealed subsets of co-regulated genes, allowing Shin and his colleagues to identify gene subnetworks that either promote or constrain differentiation. These were connected via the MYBgene, which acts as a ‘hinge’ that can potentially trigger the activation of either subnetwork.
Shin hopes to extend the same kind of analysis to the entire cellular cohort of genes, but his team’s success in characterizing expression dynamics in cancer cells also suggests clinical applications. “We wish to study this fascinating biological phenomenon in multiple cancer cell lines in response to various drugs now used in clinics,” says Shin. “This will reveal the causal factors for variation in cellular response, which can be used to improve targeted treatment.”
References
- 1. Kouno, T., de Hoon, M., Mar, J. C., Tomaru, Y., Kawano, M., Carninci, P., Suzuki, H., Hayashizaki, Y. & Shin, J. W. Temporal dynamics and transcriptional control using single-cell gene expression analysis. Genome Biology 14, R118 (2013). doi: 10.1186/gb-2013-14-10-r118