Oct. 31, 2024 Research Highlight Biology
Decoding protein interactions in cells
The ‘grammar’ governing how biomolecules form, and how cells organize themselves, is captured by a new mathematical model
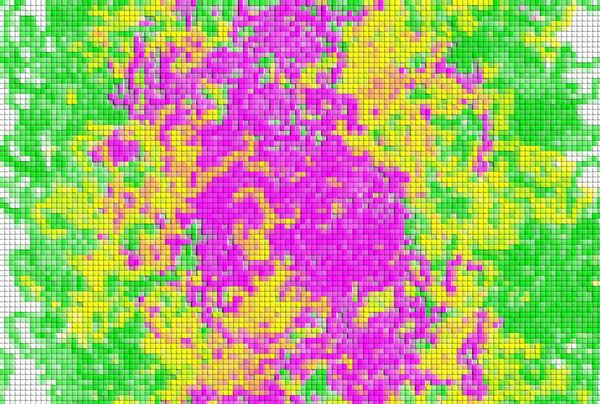
Figure 1: An example simulation of demixing of proteins. The different colors indicate different components of proteins. © 2024 RIKEN Center for Biosystems Dynamics Research
Biological cells are incredibly complex mixes of proteins, nucleic acids, lipids and carbohydrates. A model developed by two RIKEN researchers helps explain why components of the same type sometimes do and sometimes don’t spontaneously come together to form tiny droplets known as condensates1.
“Cells contain multiple condensates, which are organelles that aren’t encased by membranes,” explains Kyogo Kawaguchi of the RIKEN Center for Biosystems Dynamics Research (BDR).
Condensates form spontaneously through a process called phase separation. “Think of it like oil separating from water in salad dressing,” says Kawaguchi.
Condensates are crucial because they help organize the cell by concentrating specific molecules in the same region. They also create hubs for vital cellular processes such as gene regulation, stress response and signal transduction. Without condensates, these processes would be less efficient or even dysfunctional.
Regions in protein sequences that are intrinsically disordered are known to play an important role in condensate formation. These regions have been extensively studied, but precisely how they participate in condensate formation hasn’t been clear until now.
This process of coexisting types of condensates spontaneously separating from each other is known as demixing (Fig. 1). These demixed condensates do not merge or mix even when they happen to collide.
The opposite of demixing is hypermixing: different components stay mixed within a single condensate because they prefer to stay this way rather than be just by themselves.
“The key question we wanted to address was whether we could predict whether two or more disordered protein sequences will demix or hypermix based on the order of their amino acids—the organic building blocks of proteins,” says Kawaguchi. “We were looking for a rulebook that governs how protein sequence dictates condensate behavior.”
Now, Kawaguchi and BDR colleague Kyosuke Adachi have modeled condensate formation in more than 200 intrinsically disordered regions found in human proteins. They did this by conducting molecular-dynamics simulations.
“Molecular-dynamics simulations use computers to mimic the movements and interactions of atoms and molecules,” explains Kawaguchi. “They allowed us to study how molecules behave over time—how they interact with each other, and what structures they form.”
Using this data, the pair developed a theoretical framework that uses the information about the interactions between amino acids to predict the tendency of sequences to demix or hypermix. They then fine-tuned their framework until they could estimate the outcome of the simulation even before running it.
“We validated our theory by designing new protein sequences that successfully demixed from a given sequence in a simulation, which demonstrated its predictive power.”
“The next step is to test our predictions experimentally, validating our theory in the real world,” says Kawaguchi.
Related content
- Supercomputer simulations reveal how protein crowding in cells impacts interactions
- Uncovering the cell’s read–write mechanism for gene expression instructions
- Two-part tango triggers signal activation of key regulatory protein
Rate this article
Reference
- 1. Adachi, K. & Kawaguchi, K. Predicting heteropolymer interactions: Demixing and hypermixing of disordered protein sequences. Physical Review X 14, 4902 (2024). doi: 10.1103/PhysRevX.14.031011