2010年1月29日
独立行政法人 理化学研究所
分子1つ1つの運動まで再現する細胞シミュレーション法を開発
-細胞丸ごと規模の超精密な世界初のシミュレーション実現へ-
ポイント
- 新たな細胞内の情報伝達機構を発見し、従来の理論に拡張を迫る
- 国産の細胞シミュレーターE-Cellに搭載し、次世代スーパーコンピュータで活用拡大
- 細胞のがん化や幹細胞の分化の予測と制御などに貢献
要旨
独立行政法人理化学研究所(野依良治理事長)は、細胞内に存在する分子1つ1つの運動まで精密に再現する革新的な細胞シミュレーションの要素技術の開発に成功し、従来の理論に拡張を迫る発見をしました。これは、理研基幹研究所(玉尾皓平所長)生化学シミュレーション研究チームの高橋恒一チームリーダーとオランダの原子分子物理研究所のピーターレイン・テンウォルデ教授らとの共同研究の成果です。開発した新技術は、国産の細胞シミュレーターE-Cell※1の次世代版に搭載し、神戸に建設中の次世代スーパーコンピュータで活用し、複雑な機能を持ち、生命活動の根幹となる細胞を丸ごとシミュレーションすることを目指します。
従来の細胞シミュレーションでは、細胞内の分子1つ1つの運動を考慮せず、分子間の相互作用を回路図のように表現する手法が主流でした。しかし、生体機能を正確に理解するためには、細胞内の分子の動きやゆらぎを知ることは欠かせません。これまでもその重要性は認識されていましたが、膨大な計算時間が必要となるため、シミュレーションは現実的ではありませんでした。研究グループは、新たに超高性能の計算手法「enhanced Greens Function Reaction Dynamics(eGFRD)法」を開発し、これまで年単位で必要だった計算時間を数日以内など飛躍的に短縮することに成功しました。これにより、細胞丸ごとの規模で、1つ1つの分子の動きを精密に追ったシミュレーションを世界で初めて実現することになります。
研究グループは、新たに開発した計算手法を、細胞の意思決定にかかわる重要なタンパク分子である「MAPキナーゼ※2」のシミュレーションに適用しました。その結果、MAPキナーゼの情報伝達様式の発生条件が、現在一般的に用いられている理論が予測していたものと大幅に異なる場合があることを見いだし、理論を拡張する必要があることを発見しました。
この新たな計算手法を次世代スーパーコンピュータに搭載し、さらに発展させることで、細胞のがん化の機序の解明や、幹細胞(万能細胞)の分化の予測と制御を通じた再生医療の実現に貢献することが期待できます。さらに、有用微生物の設計に用いると、環境問題やエネルギー問題の解決の糸口となる可能性もあります。
本研究成果は、米国科学アカデミー紀要『Proceedings of the National Academy of Sciences of the United States of America: PNAS』2月9日号に掲載されるに先立ち、1月25日の週にオンライン掲載されました。
背景
微生物からヒトまで、すべての生命活動は、細胞内の生体分子同士の反応の連続である「生化学ネットワーク」の働きによって引き起こされています。生化学ネットワークの挙動を精密に模擬、予測する細胞シミュレーションは、生命活動を正確に理解し、応用へと発展していくために重要な基盤技術といえます。従来の細胞シミュレーションでは、細胞内に存在する分子と分子の相互作用を回路図のように表現し、分子の数の増減だけを扱う「ネットワークモデル」や、細胞内の場所による平均的な濃度の違いを表現する「濃度平均場モデル」といった手法が主流でした。その一方、近年の蛍光顕微鏡などの観察技術の急速な発展で、1分子単位のゆらぎや運動が生命機能に重要な役割を果たしていることが明らかになってきました。このため、生体分子1つ1つの運動まで考慮に入れて細胞の働きを精密にシミュレーションする、「1分子粒度」の手法が求められていました。しかし、従来の1分子粒度の手法である「ブラウン動力学」を用いて、細胞丸ごとの規模で精度よくシミュレーションを行うには、ごく簡単な場合でも年単位の膨大な計算量が必要でした。逆に現実的な時間で計算を終えようとすると、大きな計算誤差を許容することとなり、不正確なシミュレーションを行ってしまうのが問題でした。このため、まったく新しい技術の開発が期待されていました。
研究手法
研究グループは、細胞丸ごと規模での1分子粒度モデルを実現するために、超高性能の粒子反応拡散計算手法「enhanced Greens Function Reaction Dynamics(eGFRD)法」を開発しました。この手法は、ブラウン動力学法よりも飛躍的に性能が向上するだけでなく、原理的には系統誤差が生じないという優れた特性を持ちます。
従来の手法では、細胞内のすべての分子を少しずつ動かし、そのたびに分子と分子の衝突が発生するかどうかを判定し、化学反応をシミュレーションしていました(図1左)。この時、どの位置、どの時点で分子の衝突が起こるのかをいかに精密に計算できるかがシミュレーションの精度と直接関係するため、小さな時間間隔で非常に多くの計算を繰り返す必要がありました。開発したeGFRD法では、分子を少しずつ動かすのではなく、ある分子がほかの分子に邪魔されることなく自由に動ける範囲「保護領域」を設定し、分子が保護領域に入って何秒後に抜け出るかを計算します(図1右)。この計算を、シミュレーションを実施する多数の分子に別々に(非同期に)適用します。また、2つの分子が近くにある時は、分子が2つ入った保護領域の中で、分子の運動と反応を一度に計算するようにしました。これにより、一度に進めることができる時間間隔を拡大させることができ、計算量が大幅に減り、飛躍的な計算時間の短縮につながりました。また、分子が保護領域に入って出るまでにかかる時間(第一通過時間)や反応が起きるまでにかかる時間を、基礎物理方程式の厳密解を直接用いて計算するため、原理的には系統誤差の無いシミュレーションが可能になり、シミュレーションの精度も大幅に向上しました。
研究成果
研究グループは、開発したeGFRD法をMAPキナーゼ(MAPK)のシミュレーションに適用しました。MAPKは、細胞の表面で受容した情報を適切に処理し、細胞核内の遺伝子発現機構に引き渡す役割を持ち、細胞の意思決定にかかわる重要な細胞内情報処理分子です。1つのMAPK分子は2つのリン酸化部位を持ち、その両方のリン酸化部位がMAPキナーゼキナーゼ(MAPKK)という酵素分子によってリン酸化(二重リン酸化)されると活性化し、下流に情報を伝えます。MAPKが情報を伝達する際には、主に「段階的応答」、「超敏感応答」、「二重安定応答」の3つの応答様式のいずれかで行うと考えられています。段階的応答では、細胞の表面にある受容体分子が受け取った信号をそのまま遺伝子発現系に受け渡し、忠実性の高い情報処理を行います。超敏感応答では、受け取る信号から雑音を取り除き、オンかオフかのデジタルな信号に変換して遺伝子発現系に渡します。二重安定応答では、デジタル化に加え、過去の信号の種類によって現在の応答を変化させるため、MAPKは記憶素子として働くことができます。
このMAPKの二重リン酸化を、従来のネットワークモデルと、今回可能になった分子1つ1つの運動を考慮する1分子粒度モデルの2つの手法で計算し、結果を比較しました(図2)。すると、従来のネットワークモデルが二重安定応答を予測するケースであっても、1分子粒度モデルではそれが消失する場合があることを発見しました。MAPKをリン酸化する酵素であるMAPKKが、1つ目のリン酸化部位をリン酸化してから、次のリン酸化部位をリン酸化することができるようになるまでには一定の時間(緩和時間)がかかります。例えば、MAPKKは、リン酸化を行うためのリン酸基をATP(アデノシン三リン酸)という分子から受け取り、リン酸基を渡したATPはADP(アデノシン二リン酸)になりますが、MAPKKが次のリン酸化を行うためには、ADPを離し新しいATPの供給を受けなければなりません。今回、精密なシミュレーションにより、緩和時間が短い場合には、二重安定応答が消失することが分かりました(図3)。また、同様に超敏感応答が段階的応答に変化する場合があることや、刺激に対するMAPKの二重リン酸化の速度がこれまでの予測よりも速く、MAPKの拡散速度に反比例する場合があることも分かりました。
従来のネットワークモデルは、生体分子1つ1つの運動を考慮せず、分子が衝突し、反応を起こす確率が均一であるとする化学マスター方程式※3という理論に基づいています。eGFRD法のような、分子1つ1つの運動も考慮した精密なシミュレーションにより、MAPKが情報伝達を行う「超敏感応答」や「二重安定応答」などの応答様式の発生条件が、これまでの理論とは大幅に異なる場合があることを発見できました。これまでMAPKの反応機構の解明には多数の研究者が取り組んでいましたが、実験結果と理論とが必ずしも一致しないことが謎でした。今回の成果によりMAPKの反応機構の解明が一層進むことが期待できます。
今後の期待
eGFRD法を用いると、MAPKなど特定の分子にターゲットを絞れば、在来の計算機でも細胞丸ごと規模でのシミュレーションが可能であることが分かりました。このeGFRD法を細胞シミュレーターE-Cellの次世代版に搭載し、次世代スーパーコンピュータで活用することで、細胞内に存在する多くの種類の分子の同時シミュレーションや、多数の細胞からなる組織のシミュレーションへ道を開くことになります。具体的には、複数の情報伝達経路同士の相互作用(クロストーク)の影響や、細胞内の分子の混み合い方(分子混雑)が及ぼす影響、また細胞同士が信号分子を交換してコミュニケーションを行う様子など、さらに高度なシミュレーションが実現します。このように精密かつ大規模な細胞シミュレーションが実現すると、最終的には、細胞のがん化の機序の解明を通じて創薬へ貢献することができたり、幹細胞(万能細胞)の分化の予測と制御を通じて再生医療の実現へとつなげることができると期待されます。さらに、有用微生物の設計に適用すると、環境問題やエネルギー問題の解決の糸口を見いだす可能性もあると考えています。
発表者
理化学研究所
基幹研究所 先端計算科学研究領域
システム計算生物学研究グループ
生化学シミュレーション研究チーム
チームリーダー 高橋 恒一(たかはし こういち)
Tel: 045-503-9430 / Fax: 045-503-9429
報道担当
理化学研究所 広報室 報道担当Tel: 048-467-9272 / Fax: 048-462-4715
補足説明
- 1.E-Cell
1996年に慶應義塾大学で発足したE-Cellプロジェクトにより開発された、国産の細胞シミュレーター。E-Cell Systemは、複数の計算法や時間スケールなどが混在しても効率的にシミュレーションできるなどの特徴を持つ統合ソフトウエアプラットフォームで、細胞のような極度に複雑な系での効果的なシミュレーションを目指す。現在、理研で次世代版バージョン4の開発が進んでいる。 - 2.MAPキナーゼ
分裂促進因子活性化タンパク質キナーゼ(Mitogen-activated Protein Kinase)の略称。MAPKとも呼ばれる。細胞が何らかの刺激を受けた時に活性化される。活性化されたMAPKは細胞核内に移行し、遺伝子の発現の制御に関与する。 - 3.化学マスター方程式
細胞内の溶液などで起きる化学反応を記述した式。例えば、細胞内に活性化前のMAPKが9個、活性化したMAPKが11個あった場合、反応が起きて活性化前後のMAPKがそれぞれ10個になる確率など、起きうるあらゆる反応の可能性を含む。このため、マスター方程式と呼ばれる。ある時点で起きる反応の確率は、分子の位置や運動によらず均一であるという仮定に基づくが、今回の研究でこの仮定が必ずしも有効ではないことが明らかになった。
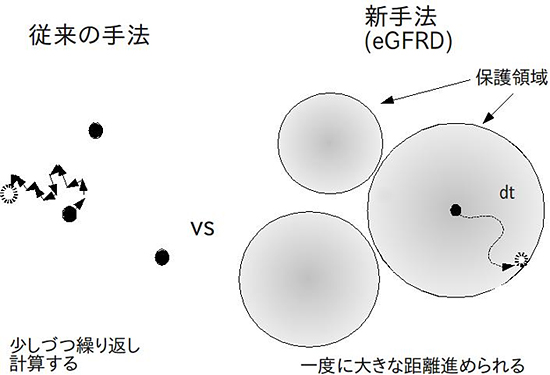
図1 従来の計算手法とeGFRD法との比較
eGFRD法では、分子が自由に運動できる保護領域を設定するため、一度に大きな距離を進められる。
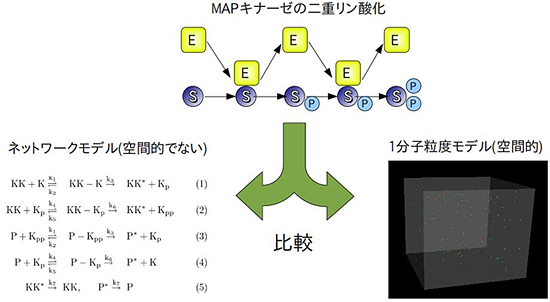
図2 MAPK系のシミュレーション
(EはMAPKK、SはMAPK、Pはリン酸基)
MAPKの二重リン酸化を、空間中の分子の運動を考慮しないネットワークモデルと、空間を考慮した1分子粒度モデルで計算し、結果を比較した。
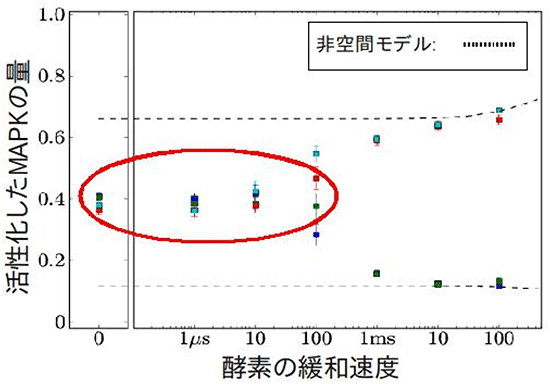
図3 MAPKシミュレーションの結果
酵素の緩和速度が速くなると、MAPKの二重安定応答が消失する場合がある(赤丸)。