2012年7月13日
独立行政法人 理化学研究所
難治性の骨疾患「短体幹症」の原因遺伝子を発見
-軟骨代謝に必要な酵素「PAPSS2」の機能喪失で短体幹症が発症-
ポイント
- 次世代シーケンサーを用いた骨の難病の原因究明プロジェクト、初の成果
- PAPSS2遺伝子の解析による1型短体幹症の遺伝子診断、保因者診断が可能に
- PAPSS2の機能解析を進め、短体幹症に対する有効な治療法の開発へ
要旨
理化学研究所(野依良治理事長)は、脊椎の異常により体幹の短縮を起こす1型短体幹症の原因遺伝子がPAPSS2遺伝子であることを発見しました。これは、理研ゲノム医科学研究センター(久保充明センター長代行)骨関節疾患研究チームの池川志郎チームリーダー、飯田有俊上級研究員と横浜市立大学環境分子医科学の三宅紀子准教授らを中心とする共同研究グループによる成果です。
短体幹症とは、脊椎の異常により体幹(胴体)の短縮を起こす疾患の総称で、その遺伝形式※1と病像により多くの疾患に分けられています。いずれの疾患も1種類の遺伝子の変異により発症する単一遺伝病で、脊柱が側方へ曲がりねじれる側彎(わん)など脊椎の変形、早期の椎間板の変性、四肢関節の異常など、多様な骨格異常を起こす難病です。そのため、発症原因の解明、予防・治療法の確立が待ち望まれています。
研究グループは、3人の患者を持つ1つの家系を詳細に検討し、常染色体劣性遺伝の遺伝形式を持つ1型短体幹症の家系であることを確認しました。次世代シーケンサー※2を用いたエキソーム解析※3という研究手法を用いてこの3人の患者のゲノムを広範囲に調べた結果、3人に共通する遺伝子変異としてPAPSS2遺伝子の1塩基の挿入変異を発見しました。PAPSS2は軟骨の代謝に重要な役割を果たす酵素として知られており、この変異はPAPSS2の機能を喪失させると考えられます。家系内でこの変異を調べると、遺伝形式に対応して変異が伝わっていることが確認できました。さらに、この家系の患者と類似の病態を示す非家族性の患者3人について、PAPSS2遺伝子の変異を調べたところ、いずれの患者にも相同染色体の両方にPAPSS2遺伝子の機能を喪失させる別の変異を同定しました。
今回、1型短体幹症の原因遺伝子を世界で初めて発見しました。今後、PAPSS2遺伝子の解析により1型短体幹症の遺伝子診断や保因者※4診断が可能になります。また、PAPSS2の機能解析を通じて、短体幹症の病態や軟骨代謝の機構の理解が進み、短体幹症やそれに類する疾患に対する有効な治療法が開発されることが期待できます。
本件は、厚生労働省難病関係研究分野のプロジェクト、『遺伝性難治疾患の網羅的エキソーム解析拠点の構築』の一環として行われたもので、英国の医学雑誌『Journal of Medical Genetics』(7月11日)に掲載されました。
背景
理研ゲノム医科学研究センター 骨関節疾患研究チーム(池川志郎チームリーダー)は、ゲノム科学の基礎研究成果を医療現場へ見える形で還元することを目指して、平成23年度より、厚生労働省難病関係研究分野のプロジェクト『遺伝性難治疾患の網羅的エキソーム解析拠点の構築』(班長:横浜市立大学環境分子医科学 松本直通教授)に参加し、遺伝性骨関節疾患のゲノム解析とそのゲノム医科学への展開に取り組んでいます。特にこれまで、理研ゲノム医科学研究センター、及び同チームで蓄積した大規模ゲノム解析のノウハウは、遺伝性疾患や難病の研究に貢献できると期待しています。
骨関節には多くの遺伝性疾患が存在します。ほとんどが単一遺伝病、すなわち1種類の遺伝子に変異があるために発症する遺伝病です。骨関節の遺伝病は、各疾患により特徴的な異常パターンを示し、そのパターンにより多くのグループに分けられています。
その中で短体幹症は、脊椎の異常により体幹(胴体)の短縮を起こす疾患のグループです。このグループには多くの疾患が属し、遺伝形式と脊椎変形の形状や合併異常の有無などの特徴により臨床的に1型から3型に分類され、更に各型が細かく分類されています(表1)。しかし、分類の基準は、非客観的かつ不明瞭で、骨関節の単一遺伝病の中でも、最も研究が遅れている疾患グループの1つです。短体幹症は、いずれの疾患も、椎体の扁平化、脊柱が側方へ曲がりねじれる側彎(わん)などの変形、早期の椎間板の変性、四肢関節の異常など骨格の異常をきたす難病で(図1)、その発症原因の解明、予防・治療法の確立が待ち望まれています。
そこで、研究グループは、最先端の大規模ゲノム解析の手法を用いて集中的にこの疾患の遺伝子解析を行うことで、早期に原因遺伝子を究明し、遺伝的診断手法を確立することに挑みました。
研究手法と成果
研究グループの中の医師、研究者が参加しているネットワーク、「骨系統疾患コンソーシアム※5」(池川志郎、西村玄代表)は、3人の短体幹症患者を持つトルコ人家系の協力を得ました。家系内での病気の伝わり方を詳細に検討したところ、常染色体劣性遺伝の遺伝形式を持つことが確認できました。また、患者の臨床像、X線画像の検討から1型短体幹症であると診断しました。
研究グループの三宅准教授らは、次世代シーケンサーを用いたエキソーム解析という研究手法を用いて、3人の患者のゲノムを広範囲に調べました。3人に共通する遺伝子の変異を調べたところ、PAPSS2遺伝子に1塩基の挿入変異を発見しました。PAPSS2遺伝子は、常染色体の10番染色体長腕上に存在する遺伝子です。PAPSS2 (phosphoadenosine-phosphosulfate synthetase 2)は、軟骨の基質の硫酸化に関係し、軟骨代謝に重要な役割を果たす酵素として知られています。今回発見した変異は、余分な塩基が1つ挿入されることで遺伝子のタンパク質の読み枠がずれてフレームシフト※6とよばれる異常を起こし、正常より極端に短いタンパク質ができるためPAPSS2の機能が喪失すると示唆されました。
次世代シーケンサー研究の大きな問題の一つは、精度が低く、得られたシーケンスに擬陽性が多いことです。そこで飯田上級研究員らは、現在標準的に用いられているサンガー・シーケンス法※7でこの変異を調べ、3人の患者がいずれも相同染色体の両方にこの変異を持っていることを確認しました。また、家系全体でこの変異を調べ、常染色体劣性の遺伝形式に対応して変異が伝わっていることが確認しました。さらに、骨系統疾患コンソーシアムの協力を得て、この家系の患者と類似の臨床像を持つ非家族性の患者3人のゲノムについて、PAPSS2遺伝子の変異を調べました。その結果、いずれの患者も相同染色体の両方にPAPSS2遺伝子の機能を喪失させるような別の変異を持つことが分かりました(表2)。
変異を持つ計6人の患者の臨床的な特徴やX線画像を検討すると、脊椎の異常は極めて類似していましたが、それ以外の四肢関節の異常や肋骨の石灰化などの症状は、家系内でもかなりの違いがあることが分かりました。従来、いくつかの症状によって1型短体幹症が細分類されていました。しかし、今回の結果から1型短体幹症発症の原因遺伝子は1つであり、報告されていたさまざまな症状は、個人差や異なる発症時期や進行度合に由来することが分かりました。
今後の展開
今回の原因遺伝子の発見により、PAPSS2遺伝子解析による1型短体幹症の遺伝子診断、保因者診断が可能になります。また、不明瞭だった短体幹症の分類の整理が進み、疾患の概念がより明確になり、臨床診断が容易になります。
今後、研究グループは、PAPSS2の機能解析を通じて短体幹症の病態を解明し、短体幹症やその類縁疾患の画期的な治療法開発を目指します。PAPSS2変異による1型短体幹症は、酵素の欠損によって引き起こされるため、早期に疾患を発見しPAPSS2を補充することによって症状の改善、治癒が期待できます。
原論文情報
- Noriko Miyake, Nursel H, Elcioglu, Aritoshi Iida, Pinar Isguven, Jin Dai, Nobuyuki Murakami, Kazuyuki Takamura, Tae-Joon Cho, Ok-Hwa Kim, Tomonobu Hasegawa, Toshiro Nagai, Hirofumi Ohashi, Gen Nishimura, Naomichi Matsumoto, and Shiro Ikegawa.
“PAPSS2 mutations cause autosomal recessive brachyolmia.”Journal of Medical Genetics(2012)DOI: 10.1136/jmedgenet-2012-101039
発表者
理化学研究所
横浜研究所
ゲノム医科学研究センター
骨関節疾患研究チーム
チームリーダー 池川 志郎(いけがわ しろう)
お問い合わせ先
横浜研究推進部 企画課
Tel: 045-503-9117 / Fax: 045-503-9113
報道担当
理化学研究所 広報室 報道担当
Tel: 048-467-9272 / Fax: 048-462-4715
補足説明
- 1.遺伝形式
遺伝(親から子への形質の伝達)のパターンのこと。遺伝子の染色体上の位置(常染色体にあるのか、性染色体にあるのか、ミトコンドリアにあるのかなど)と、変異効果の優性、劣性により、常染色体優性、常染色体劣性、X染色体優性、X染色体劣性、Y染色体劣性などがある。 - 2.次世代シーケンサー
現在、標準的に使われているSanger(サンガ―)法に基づくDNAシーケンサーに対して、それと異なる原理に基づく、より高速、大量のシーケンス能力を持つシーケンサーのこと。多くの種類のシーケンサーが開発されているが、得られるシーケンスの精度が低いのが共通の問題になっている。 - 3.エキソーム解析
ゲノムの中のタンパク質に関する情報を含むエクソン部分(ゲノム全体の約3%)を、シーケンスにより包括的に解析しようというゲノム科学の手法。 - 4.保因者
遺伝病の原因遺伝子を持っているのに発症していない人。本人は遺伝病ではないが、子供が遺伝病を発症する可能性を持つことになる。優性遺伝病の保因者は、遺伝子の不浸透(遺伝子の効果が顕在化しないこと)による。劣性遺伝病の保因者は、一対の染色体の一方にのみ原因遺伝子変異を持つ。 - 5.骨系統疾患コンソーシアム
骨関節の単一遺伝病である骨系統疾患の医療の改善・発展を目指して立ち上げられた日本のネットワーク。創設者の西村玄(都立小児総合医療センター)、池川志郎を中心として、多くの医師、基礎研究者、患者が、ボランティアとして参加・運営している私的な非営利組織。
骨系統疾患コンソーシアム - 6.フレームシフト
塩基の欠失または挿入により、遺伝子のコドンの読み枠がずれること。コドンは、3つの塩基からなるので、3の倍数でない数の塩基の欠失、または挿入によりこのタイプの変異が起こることになる。アミノ酸配列が変わり、正常とは全く異なるタンパクができることになる。 - 7.サンガー・シークエンス法
フレディック・サンガーが発見した原理(ジデオキシ法)に基づいた、現在標準的に使われているDNAシーケンスの方法。
表1 従来の短体幹症の分類
![]() 左右にスクロールできます
左右にスクロールできます
| 分類(亜型) | 遺伝形式 | 臨床的細分類 | 特徴 |
|---|---|---|---|
| 1型 | 常染色体劣性 | Hobaek型 | 細長い長方形の椎体 |
| 常染色体劣性 | Toledo型 | 角膜混濁、肋骨の早期の石灰化 | |
| 2型 | 常染色体劣性 | Maroteaux型 | 円形の椎体、大脳鎌の早期の石灰化 |
| 3型 | 常染色体優性 | TRPV4変異によるもの | 1、2型に比べ、頸椎の扁平椎や側弯が強い |
| TRPV4変異のないもの | |||
| その他 | 常染色体優性 | ||
| 常染色体劣性 | |||
| 不詳 |
亜型のその他の部分は、ほとんどが1家系、または1例のみの報告で、疾患としての明確な特徴、定義がない。
表2 1型短体幹症患者で発見されたPAPSS2遺伝子の変異
![]() 左右にスクロールできます
左右にスクロールできます
| 家系 ID |
変異の遺伝子上の位置 | DNAの変化 | 予想されるアミノ酸の変化 |
|---|---|---|---|
| 1 | Exon 3 | 337_338insG (1塩基の挿入) | A113GfsX18 |
| (homozygous) | |||
| 2 | Exon 5 | 616-634del19 (19塩基の欠失) | V206SfsX9 |
| Exon 11 | 1309-1310delAG (2塩基の欠失) | R437GfsX19 | |
| 3 | Intron 3 | IVS3+2delT (1塩基の欠失) | L50SfsX2 |
| (homozygous) | |||
| 4 | Exon 4 | 480_481insCGTA (4塩基の挿入) | K161RfsX6 |
| Exon 6 | 661delA (1塩基の欠失) | I221SfsX40 |
PAPSS2遺伝子には、計13のエクソン (ゲノムDNAの内、mRNAに転写される部分)がある。これをゲノムDNAに対するPCR-ダイレクト シーケンス法で変異を調べた。
homozygous:父母由来の染色体の両方に、同じ変異が見られることを表す。同定された6種類の変異は、フレームシフト(fs)により、いずれも早期にストップコドン (Xで表される)を生じ、正常に比べ遥かに短いPAPSS2タンパク質を生ずると示唆された。
アミノ酸の変化の読み方 例A113GfsX18:
変異により、113番目のアミノ酸A(アラニン)がG(グリシン)に代わり、以下、フレームシフト(fs)を起こして、18個目にストップコドン (X)がきて、翻訳が終わり、短いタンパク質ができてしまう。
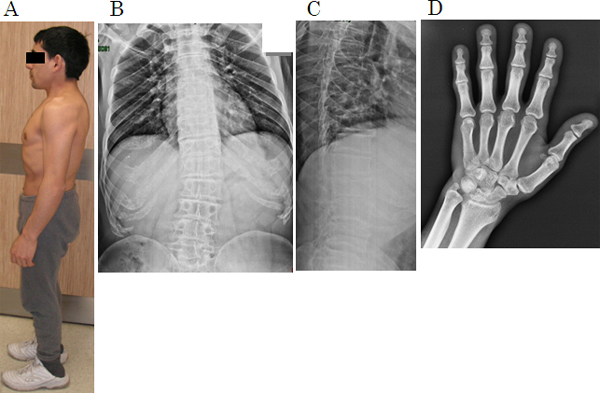
図1 1型短体幹症の病態(29歳、男性患者)
Aは外観、B~DはX線画像。
- A: 短体幹型(胴体が短くなるタイプ)の低身長(135cm)。相対的に手足が長く見える。四肢の奇形等はない。
- B: 脊椎正面像。疾患による脊椎の発達障害のために、脊椎椎体の高さが低くなっている。軽度の側弯変形、肋骨の早期石灰化も見られる。
- C: 脊椎側面像。脊椎椎体は扁平で、長方形になっている。椎間板の幅が狭くなっている。早期の椎間板変成症の像である。
- D: 左手正面像。中手骨の短縮がみられる。