要旨
理化学研究所(理研)統合生命医科学研究センター疾患システムモデリング研究グループの北野宏明グループディレクター、川上英良特別研究員らの共同研究グループは、遺伝子発現データから遺伝子制御に重要な転写因子[1]を網羅的に予測する手法を開発しました。
遺伝子発現の制御は、主にDNAの配列特異的に結合する転写因子によって行われています。そのため、制御に重要な転写因子を同定することは、疾患や正常細胞機能の解明に重要です。しかし、1,000種類以上存在するといわれる転写因子の制御活性を網羅的に計測するのはいまだに困難です。
共同研究グループは、転写因子が遺伝子のどの領域に結合しているかを網羅的に測定した、クロマチン免疫沈降オンチップ(ChIP on chip)[2]やクロマチン免疫沈降-次世代シーケンシング(ChIP-seq)[3]のデータを、世界中から3,500実験以上集めて再解析を行うことで、転写因子と標的遺伝子間の制御ネットワークである「遺伝子制御ネットワーク」を構築しました。この遺伝子制御ネットワークは、450種類以上の転写因子の結合情報に加えて、従来の転写因子の結合モチーフ配列[4]に基づく遺伝子制御ネットワークでは考慮されない“それぞれの転写因子が、実際にどのくらいの頻度で各遺伝子の制御領域に結合しているか”という情報を含んでいます。
さらに、従来のGene Set Enrichment解析[5]に確率的な制御関係を考慮したwPGSA法[6](weighted Parametric Gene Set Analysis)を導入することで、遺伝子発現データから450種類以上の転写因子の活性を極めて高い精度で予測することに成功しました。また、この手法を組織横断的な遺伝子発現データやインフルエンザウイルス感染マウスモデルの時系列遺伝子発現データに適用することで、組織や疾患に特異的な転写因子群を予測しました。
この手法は、さまざまな生命現象や疾患の制御メカニズムを明らかにする上で幅広く利用されることが期待できます。この手法の Webインターフェース(英語)はライフサイエンス統合データベースセンター(DBCLS)の支援のもと公開されており、手持ちの遺伝子発現データから転写因子の予測を行うことができます。
本研究は、文部科学省科学研究費補助金の支援のもとで行われました。
成果は、英国の科学雑誌『Nucleic Acids Research』オンライン版(4月30日付け)に掲載されました。
※共同研究グループ
理化学研究所 統合生命医科学研究センター 疾患システムモデリング研究グループ
グループディレクター 北野 宏明 (きたの ひろあき)
特別研究員 川上 英良 (かわかみ えいりょう)
東京大学 大学院医学系研究科 国際保健政策学
助教 中岡 慎治 (なかおか しんじ)
ライフサイエンス統合データベースセンター(DBCLS)
特任研究員 大田 達郎 (おおた たつろう)
背景
細胞周期、ストレス応答、代謝といった細胞の多くの機能は、遺伝子発現の結果として生じます。遺伝子発現の制御は、主にDNAの配列特異的に結合する転写因子によって行われています。そのため、制御に重要な転写因子を同定することは、疾患や正常細胞機能を解明する第一歩として重要です。
近年、転写因子をテーマとした多くの研究が行われていますが、1,000種類以上存在するといわれる転写因子の制御活性を網羅的に計測するのはいまだに困難です。転写因子の活性は、そのタンパク質の量だけではなく、リン酸化、糖鎖付加、アセチル化といった修飾状態、複合体形成、細胞内輸送などさまざまな要因によって翻訳後調節を受けています。よって、これらを毎回多くの転写因子について、網羅的に測定するのは非現実的でした。
一方、マイクロアレイ法[7]や次世代シーケンシング技術[8]の普及によって、遺伝子発現を網羅的に計測することは比較的容易になってきています。したがって、転写因子とその制御対象である遺伝子との間を適切な関係性(ネットワーク)でつなぐことができれば、計測が容易な遺伝子発現の変化から転写因子の状態を予測し、重要な転写因子を同定することができると考えられます(図1)。
研究手法と成果
今回、共同研究グループは、近年膨大に集積しつつある世界中のクロマチン免疫沈降オンチップ(ChIP on chip)やクロマチン免疫沈降-次世代シーケンシング(ChIP-seq)といった、転写因子の結合を網羅的に計測したビッグデータを用いることで、転写因子と標的遺伝子間の制御関係である「遺伝子制御ネットワーク」を構築しました。
これまでも、転写因子の結合モチーフ配列に基づく遺伝子制御ネットワークはありましたが、
①転写因子は配列特異的に結合するものの結合頻度にかなりの差がある
②結合モチーフが分かっていない転写因子が多い
③結合モチーフが似ている転写因子は区別がつきにくい
といった問題点がありました。
共同研究グループは、さまざまな条件で計測された転写因子の遺伝子への結合データを統計的に活用することで、「それぞれの転写因子が、実際にどのくらいの頻度で各遺伝子の制御領域に結合しているか」という情報を含んだネットワークを構築しました(図2)。
Gene Set Enrichment解析は、特徴的な発現変化をしている遺伝子集団を検出する手法として幅広く使われています。共同研究グループは、転写因子の結合頻度の情報を活用するために、従来のGene Set Enrichment解析に“重み”の概念を導入した、wPGSA(weighted Parametric Gene Set Analysis)法を考案しました。この手法の導入により“重み”を考慮しない従来の手法より高い信頼性で生物学的な知見に合致した、450種類以上の転写因子の活性を予測することに成功しました。
wPGSA法を用いた転写因子活性予測は、マイクロアレイ法や次世代シーケンシング技術で得られたあらゆる遺伝子発現変動データに適用可能であることを特長とします。
実際に、インフルエンザウイルス感染のマウスモデルにおける時系列遺伝子発現データ注1)を対象にwPGSA法を適用したところ、低病原性インフルエンザウイルスと高病原性インフルエンザウイルスがマウスにおいて著しく異なる転写因子の活性化を誘導することが明らかになりました(図3)。低病原性ウイルスは、感染初期に多くの転写因子を活性化するのに対し、高病原性インフルエンザウイルス感染では、初期に抑制されている炎症応答転写因子(STAT、IRF、NFKBなど)が感染後18時間以降に急激に活性化することが分かりました。さらに、「P-TEFb」複合体がこれらの炎症応答転写因子を制御していることが示唆され、今後抗インフルエンザウイルスの創薬ターゲットとして期待されます。
注1)Shoemaker JE.et al. PLoS Pathog 11 (6)(2015)
今後の期待
本手法を用いてさまざまな遺伝子発現データから、疾患や細胞応答に重要な転写因子を予測することが可能になりました。疾患や細胞応答において、今まで報告されていない転写因子の関与が明らかになり、制御メカニズムの解明を促進すると考えられます。この手法のWebインターフェース(英語)はライフサイエンス統合データベースセンター(DBCLS)の支援のもと公開されており、手持ちの遺伝子発現データから転写因子の予測を行うことができます。
また、本研究ではChIPビッグデータを転写因子結合の頻度という“重み”として取り入れましたが、wPGSA法は蓄積しつつある他のオミクスビッグデータ[9]も同様に活用するフレームワークとして機能すると期待できます。
原論文情報
- Eiryo Kawakami, Shinji Nakaoka, Tazro Ohta, and Hiroaki Kitano, "Weighted Enrichment Method for Prediction of Transcription Regulators from Transcriptome and Global Chromatin Immunoprecipitation data", Nucleic Acids Research, doi: 10.1093/nar/gkw355
発表者
理化学研究所
統合生命医科学研究センター 疾患システムモデリング研究グループ
グループディレクター 北野 宏明 (きたの ひろあき)
特別研究員 川上 英良 (かわかみ えいりょう)
 北野 宏明
北野 宏明
 川上 英良
川上 英良
報道担当
理化学研究所 広報室 報道担当Tel: 048-467-9272 / Fax: 048-462-4715
補足説明
- 1.転写因子
DNAに配列特異的に結合するタンパク質で、プロモーターやエンハンサーといった転写制御領域に結合し、RNAポリメラーゼによる遺伝子の転写を活性化あるいは不活性化する。 - 2.クロマチン免疫沈降オンチップ(ChIP on chip)
転写因子に対する抗体で免疫沈降を行い、一緒に沈降してきたDNA配列をガラススライドマイクロアレイ(chip)により検出する手法。 - 3.クロマチン免疫沈降-次世代シーケンシング(ChIP-seq)
転写因子に対する抗体で免疫沈降を行い、一緒に沈降してきたDNA配列を次世代シーケンシングにより網羅的に決定する手法。 - 4.転写因子の結合モチーフ配列
転写因子が結合しやすい特異的な5~30塩基程度のDNA配列のこと。 - 5.Gene Set Enrichment解析
遺伝子の発現変動データから、特徴的な変動を示す遺伝子群(gene set)を検出する手法。2005年に『 PNAS』で発表されたGSEA法をはじめ、さまざまな統計手法が遺伝子群検出に適用されてきている。検定を用いて、特定の遺伝子群の発現変動分布と全遺伝子の発現変動分布を比較することで、特徴的な変動を示す遺伝子群を検出するparametric gene set analysisが近年提案されている。 - 6.wPGSA法
本研究において考案されたparametric gene set analysisに、重み付き検定を導入することで、転写因子の遺伝子ごとの制御頻度情報を考慮した解析手法。wPGSAとはweighted Parametric Gene Set Analysisの略。 - 7.マイクロアレイ法
塩基配列が既知の多種類の遺伝子のDNAをプローブとして、プレート上に規則正しく貼付けておく。調べたい細胞からmRNAを採取し、蛍光標識する。これをプレート上に貼付けられたDNAと反応させ、蛍光強度を読み取ることにより、mRNAの発現量を網羅的かつ定量的に調べることができる。 - 8.次世代シーケシング技術
従来のサンガー シーケンシング法を利用した蛍光キャピラリーシーケンサーである「第1世代シーケンサー技術」と対比させて使われている。解析する対象とするDNAを細かく断片化し、それらを同時並行で解析し、数千万カ所という大量の配列を読み取ることができるDNA配列解析技術。 - 9.オミクスビッグデータ
オミクスとは遺伝子、タンパク質、脂質など生命の構成因子について、網羅的に計測することを指す。近年、計測技術の向上・低価格化により、さまざまな実験条件において取得されたオミクスデータが蓄積されており、ビッグデータとしての解析・活用が求められている。
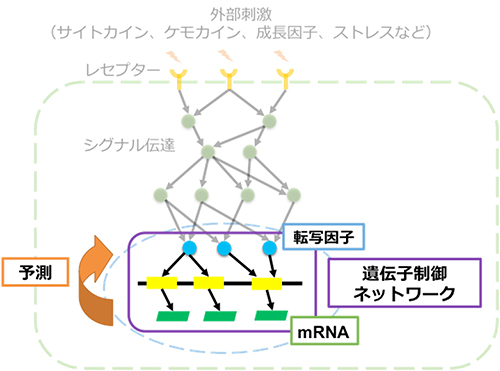
図1 細胞内の遺伝子制御ネットワーク
細胞は外部からの刺激(サイトカイン、ケモカイン、成長因子、ストレスなど)をレセプター(受容体)から受け取る。続いてシグナル伝達によって核内の転写因子に刺激情報を伝え、2~3万個の遺伝子の発現を制御することでさまざまな応答を起こす。遺伝子制御ネットワークが構築できれば、遺伝子発現変化から転写因子の状態を予測し、重要な転写因子を同定することができる。
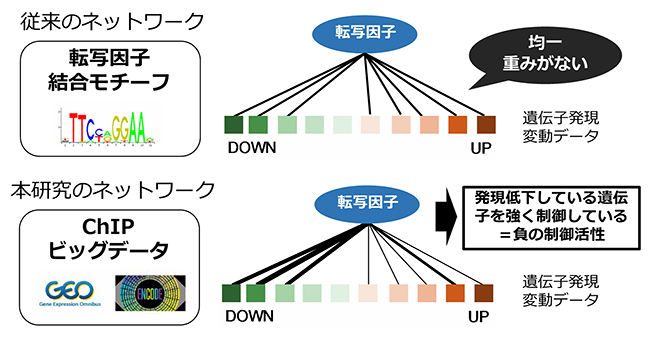
図2 従来のネットワークとChIPビッグデータを取り入れたネットワーク
従来の遺伝子制御ネットワークは結合モチーフ配列に基づくもので、転写因子の結合頻度の差を考慮できていない、結合モチーフが分かっていない転写因子が多い、結合モチーフが似ている転写因子は区別がつきにくいなどの問題があった。本研究のChIPビッグデータを取り入れた遺伝子制御ネットワークには、従来のものには含まれない結合頻度情報を含んでおり、転写因子活性の推定に適している
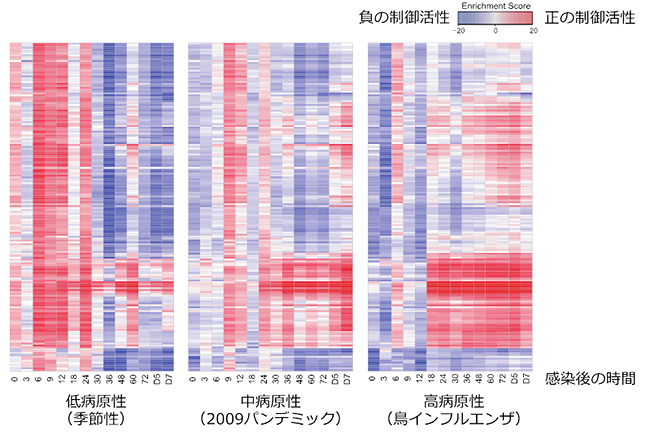
図3 インフルエンザウイルス感染マウスにおける経時的転写因子活性変化
wPGSA法によって、インフルエンザウイルス感染マウスの肺の遺伝子発現変化から、転写因子の活性予測を行った。縦軸は転写因子の種類を示す。低病原性(季節性)ウイルスは感染初期に多くの転写因子を活性化した。高病原性(鳥インフルエンザ)ウイルス感染では、初期に抑制される転写因子(炎症応答)が感染後18時間以降に急激に活性化することが分かる。中病原性(2009年新型インフルエンザパンデミック)ウイルス感染は、低病原性と高病原性の中間のような変化を示した。