要旨
理化学研究所(理研)計算科学研究機構粒子系生物物理研究チームの杉田有治チームリーダー、ジェウン・ジョン研究員、杉田理論分子科学研究室の森貴治研究員らの共同研究チーム※は、生体分子の運動を1分子レベルから細胞レベルまでの幅広い空間スケールで解析可能なシミュレーションソフトウェア「GENESIS」を開発し、5月8日からオープンソースソフトウェアとして無償で公開します。
近年、計算機によるシミュレーションは、実験、理論に次ぐ第3の解析手法として、さまざまな分野で活用されています。生命科学では分子動力学法[1]と呼ばれるシミュレーション技法が、タンパク質の立体構造予測や、酵素反応のメカニズムの解明、薬の理論設計などに広く応用されています。分子動力学法は粒子間相互作用[2]をクーロンの法則などの物理法則に基づいて計算し、ニュートンの運動方程式F = maを解くことで分子の動きをコンピュータ内で仮想的に再現する方法です。この手法を用いることで、分子集団系のピコ秒~マイクロ秒(1兆分の1秒~100万分の1秒)の時間変化を原子レベルの解像度で観察できます。
生命科学、特に分子生物学では、タンパク質や核酸(DNAやRNA)などの生体高分子が細胞という環境の中でどのように機能しているかを理解することが必要です。これまでに世界中でさまざまな分子動力学シミュレーションソフトウェアが開発されてきましたが、多数の演算装置(CPU)を用いて、従来の計算アルゴリズムを大規模な分子集団系に対して適用しようとすると、CPU間の通信時間が増大するため限界がありました。実際、タンパク質1分子の計算は可能でしたが、細胞質のように多数のタンパク質や核酸などが混在するシステムを、水やイオンなどの溶媒も含めて高速に計算することは困難でした。
共同研究チームが開発した超並列分子動力学計算ソフトウェアGENESISは、スーパーコンピュータ「京(けい)」[3]のアーキテクチャ(基本設計)を考慮に入れた独自の計算アルゴリズムを導入することで、並列計算を高効率化し、細胞環境を想定した1億個の原子で構成される系に対しても高速な分子動力学シミュレーションを実現しました。また、従来のようにタンパク質1分子や細胞膜、糖鎖、核酸などの生体分子のシミュレーションも可能であり、今後、創薬研究などに幅広く適用されると期待できます。GENESISは、GPLv2ライセンス[4]に基づくオープンソースソフトウェアとして5月8日から粒子系生物物理研究チームのホームページ※で無償で公開します。
本研究は、オンラインジャーナル『WIREs Computational Molecular Science』のオープンアクセス版(5月7日付け:日本時間5月8日)に掲載されます。
※共同研究チーム
理化学研究所
計算科学研究機構 研究部門 粒子系生物物理研究チーム
チームリーダー 杉田 有治(すぎた ゆうじ)
研究員 Jaewoon Jung (ジェウン・ジョン)
研究員 小林 千草(こばやし ちぐさ)
研究員 松永 康佑(まつなが やすひろ)
杉田理論分子科学研究室
研究員 森 貴治(もり たかはる)
長浜バイオ大学 バイオサイエンス学部
准教授 依田 隆夫(よだ たかお)
米国ミシガン州立大学 生化学分子生物学科
教授 Michael Feig(マイケル・ファイグ)
(理研生命システム研究センター 分子機能シミュレーション研究チーム 客員研究員)
背景
近年、計算機によるシミュレーションは、実験、理論に次ぐ第3の解析手法として、さまざまな分野で活用されています。生命科学では分子動力学法と呼ばれるシミュレーション技法が、タンパク質の立体構造予測や、酵素反応のメカニズムの解明、薬の理論設計などに広く応用されています。これまでに世界中でさまざまな分子動力学シミュレーションソフトウェアが開発されてきましたが、多数の演算装置(CPU)を用いて、従来の計算アルゴリズムを大規模な分子集団系に対して適用しようとすると、CPU間の通信時間が増大するため限界がありました。
実際、タンパク質1分子などの計算は可能でしたが、細胞質のように多数のタンパク質や核酸(DNAやRNA)などが混在するような生体分子システムを、水やイオンなどの溶媒も含めて、高速に計算することは困難でした。特に、スーパーコンピュータ「京」のように非常に多くのCPUを備えたコンピュータの性能を最大限に利用するためには、新しい計算アルゴリズムを導入した分子動力学シミュレーションソフトウェアの開発が必要でした。
研究手法と成果
分子集団系に関する相互作用エネルギーは、クーロンの法則などの物理法則に基づく ポテンシャルエネルギー関数(分子力場)[5]を用いて計算できます。 図1に示すように、分子集団系中には、 結合性相互作用と非結合性相互作用[6]が働いていると考えます。分子動力学法では各原子に対してこれらの相互作用によって生じる力( F)を計算して、ニュートンの運動方程式 F = maを解くことで分子の動きをコンピュータ内で仮想的に再現できます。この手法を用いることで、比較的小さな分子集団系におけるマイクロ秒(1マイクロ秒は100万分の1秒)より速い分子運動を原子レベルの解像度で観察できます。
共同研究チームが開発した「GENESIS(GENeralized Ensemble SImulation System)」は、タンパク質や核酸、生体膜、糖鎖などの生体分子だけでなく、その集合体の分子動力学シミュレーションを行います。分子動力学シミュレーションで計算に最も時間がかかるのは、分子力場に含まれる非結合性相互作用の計算です。この計算を効率的に行うために、これまで、さまざまな試みが行われてきました。その中には、分子動力学専用コンピュータ[7]を開発して、長時間の分子動力学計算を実行するというアプローチも含まれます。分子動力学専用コンピュータを用いることでミリ秒(1,000分の1秒)に至る長時間の分子運動の解析が可能になりましたが、計算できる粒子数には制限がありました。一方で、GENESISの開発目的は非常に多くの粒子を含む分子集団系の分子動力学計算を、「京」などの汎用スーパーコンピュータを用いて効率的に行うことでした。「京」など、近年開発されたスーパーコンピュータの特徴は、数万もの演算装置(CPU)が高速なネットワークによって接続されていることです。スーパーコンピュータを有効に活用するためには、全体の計算をそれぞれのCPUに分散させて処理し、CPU間のデータ通信を最少にします。この計算を行うためには、並列計算ができるプログラムを用いる必要があります。「京」のような非常に多くのCPUを用いた並列計算を行う際には、MPIとOpenMPという2つの異なるプロトコルを組み合わせたハイブリッド並列[8]を用いることが有効です。GENESISの開発ではあらかじめハイブリッド並列が考慮されています。
GENESISは、2つの分子動力学プログラム(ATDYNとSPDYN)と解析ツールで構成しています。ATDYNは粒子間相互作用の全ペアの計算を並列化するアルゴリズム(原子分割法)を用いており、SPDYNは空間全体を小さなドメイン(領域)およびセルに分割し、ドメインとその周囲のセルに含まれる粒子間相互作用ペアの計算を並列化するアルゴリズム(空間分割法)を用いています。ATDYNは小さなペプチドを対象としたシミュレーションや粗視化(そしか)分子モデルを用いた計算、あるいは新しい計算アルゴリズムの開発やテストに用いることができます。一方、SPDYNで用いた空間分割法(図2)では、データ通信が主に隣接するドメイン間で発生するためデータ通信量を大幅に削減でき、原子分割法と比べて大きな系に対して高速に計算できます。そのため、SPDYNは膜タンパク質や細胞環境などの計算を全原子モデルで計算することを想定しています。また、新規アルゴリズムとして、①Inverse Lookup Table法、②Midpoint cell法、③Volumetric decomposition FFT 法などを導入しています。これらの新規アルゴリズムを同時に用いることで、バクテリアの細胞質分子混雑環境を模倣した約1,170万個の原子を含む分子集団系に対して1日あたり17.5ナノ秒(ns)(1ナノ秒は10億分の1秒)、約1億370万個の原子を含む分子集団系に対しては1日あたり6.5nsという性能を、「京」を用いて達成しました(図3)。
GENESISのもう1つの特徴は、レプリカ交換分子動力学法(REMD法)[9]などの効率の良いアルゴリズムを導入していることです。REMD法では、対象とする分子集団系のコピー(レプリカ)を複数用意し、それぞれ異なる温度を含む分子動力学シミュレーションを並列に実行し、ある頻度でパラメータを交換することで異なる条件のシミュレーションを混合させます(図4)。このパラメータ交換により、例えば、低温から高温に至るさまざまな温度でのタンパク質の立体構造を、一度に予測することができます。また、低温では初期構造に近い状態に留まることがよくありますが、高温のシミュレーションを混合させることで初期構造とは異なる立体構造を探索できます。GENESISに導入しているREMD法は温度だけでなく圧力や表面張力、拘束力などの多様な条件を交換できます。こうした効率の良いアルゴリズムは世界中の研究者によって開発されていますが、GENESISは新規アルゴリズム開発を行うためのプラットフォームとしても用いることが可能です。
今後の期待
生体分子系の分子動力学シミュレーションは、分子動力学専用コンピュータや大規模な汎用スーパーコンピュータなどの機能向上によって、生命科学における重要性を増しています。さらに大規模な生体分子集合体に関して、より長い時間の分子運動を計算することが可能になれば、さまざまな実験データと直接比較することが可能になり、細胞内外で働くタンパク質などの生体分子の機能を原子レベルの解像度で詳しく理解し、制御できるようになるかもしれません。それは生命科学としての重要性だけでなく、創薬応用を考えた場合に直接役立つツールの1つとして利用できます。GENESISは「京」だけでなく、ワークステーションやクラスター計算機においても実行でき、GPLv2ライセンスに基づくオープンソースソフトウェアとして5月8日から粒子系生物物理研究チームのホームページ※で公開します。GENESISが分子動力学シミュレーションを行うためのソフトウェアとして、開発グループのみならず、大学や研究機関、企業などで幅広く利用されることで、生命科学の基礎研究や創薬応用の現場で役立つと期待できます。
原論文情報
- Jaewoon Jung*, Takaharu Mori*, Chigusa Kobayashi, Yasuhiro Matsunaga, Takao Yoda, Michael Feig, and Yuji Sugita (*Equally contributed), "GENESIS: A hybrid-parallel and multi-scale molecular dynamics simulator with enhanced sampling algorithms for biomolecular and cellular simulations.", WIREs Computational Molecular Science, doi: 10.1002/wcms.1220
発表者
理化学研究所
計算科学研究機構 研究部門 粒子系生物物理研究チーム
チームリーダー 杉田 有治(すぎた ゆうじ)
研究員 Jaewoon Jung(ジェウン・ジョン)
主任研究員研究室 杉田理論分子科学研究室
研究員 森 貴治(もり たかはる)
お問い合わせ先
理化学研究所 計算科学研究機構 広報国際室
担当 岡田 昭彦(おかだ あきひこ)
Tel: 078-940-5625 / Fax: 078-304-4964
aics-koho [at] riken.jp(※[at]は@に置き換えてください。)
報道担当
理化学研究所 広報室 報道担当
Tel: 048-467-9272 / Fax: 048-462-4715
補足説明
- 1.分子動力学法
分子科学、材料科学、生命科学分野におけるコンピュータ・シミュレーション法の1つ。コンピュータを用いて分子集団系内の原子間相互作用を計算し、ニュートンの運動方程式を数値的に解くことで、コンピュータ内で分子の運動を仮想的に再現する方法。ピコ秒~マイクロ秒(1兆分の1秒~100万分の1秒)オーダーで起こる現象を原子レベルで観察することができ、分子集団系の振る舞いの理解に広く用いられる。系の温度や圧力を一定に保つ方法なども開発され、実験条件に近い状況をシミュレーションで設定できることから、計算機実験とも呼ばれる。 - 2.粒子間相互作用
分子中の原子は絶えず振動運動をしており、共有結合を介して隣の原子や、さらにその隣の原子にも振動が伝わる。電気陰性度に大きな差がある原子同士が共有結合を形成すると、電子に偏りが生じ、原子は部分電荷を持つ。またイオンは正または負の電荷を持っている。そのため、原子間で静電的相互作用(クーロン力)が発生する。また、分子内の電子分布の瞬間的な非対称性や、原子周囲の環境によって分極が生じ、それらが分子間力(ファンデルワールス相互作用)を生じさせる。 - 3.スーパーコンピュータ「京(けい)」
文部科学省が推進する「革新的ハイパフォーマンス・コンピューティング・インフラ(HPCI)の構築」プログラムの中核システムとして、理研と富士通が共同で開発を行い、2012年9月に共用を開始した計算速度10ペタフロップス級のスーパーコンピュータ。 - 4.GPLv2ライセンス
GPLv2ライセンスでは、第三者によるソースコードの改変、商業利用が可能であるが、改変したソフトウェアを配布する際、改変部分を含むソースコードを公開する義務がある。 - 5.ポテンシャルエネルギー関数(分子力場)
原子・分子に働く相互作用エネルギーを計算するためには、本来、量子化学に基づく電子状態理論を用いることが必要である。しかし、電子状態理論を直接用いた場合には比較的少数の原子系の相互作用エネルギーしか計算できないため、結合性相互作用や非結合性相互作用に関する経験的関数をあらかじめ用意し、電子状態理論や実験データを再現するようにパラメータを決定することで作られた分子力場が、分子動力学シミュレーション法ではよく用いられる。生体分子系の分子力場の基礎を確立した米国の研究者(Martin Karplus, Michael Levitt, Arieh Warshel)に2013年ノーベル化学賞が授与された。 - 6.結合性相互作用と非結合性相互作用
結合性相互作用は、原子の振動運動が共有結合を介して分子内で伝播したり、共有結合が回転したときに立体障害により原子間に働く力。非結合性相互作用は、分子内の3結合以上離れた原子同士の相互作用、および分子間で働く力である。相互作用は2体間で計算し、分子集団系内にある全ての相互作用ペアを考慮する。非結合性相互作用に関しては、原子同士が遠く離れるほど相互作用はゼロに近づくため、あるカットオフ距離を設定し、それ以上離れた原子間の相互作用は計算しないようにして、計算時間を短縮する。 - 7.分子動力学専用コンピュータ
分子動力学専用の演算装置を持つ専用コンピュータ。分子動力学計算におけるボトルネックの演算のみを処理する専用演算器を備えて、高速ネットワークで接続することで、汎用コンピュータを用いた分子動力学計算と比較して100倍~1,000倍の高速化を実現した分子動力学専用コンピュータの開発が行われている。理研では生命システム研究センターの泰地真弘人副センター長らによるMDGRAPE-3/MDGRAPE-4などの開発が行われた。また、米国D.E.Shaw Researchでは、Anton/Anton 2などの開発が行われ、比較的小さな生体分子系のシミュレーションにおいて大きな成功をおさめた。 - 8.MPI/OpenMPを組み合わせたハイブリッド並列
分散メモリ型並列と共有メモリ型並列を組み合わせた並列コンピューティング技法。MPI(Message Passing Interface)を用いた並列計算では、配列変数に関して演算を行う際、MPIプロセス数に応じて配列を分割し、各プロセスが、与えられた配列部分の演算を別々に処理する。OpenMPでは、同様に配列を分割して演算処理を行うが、配列データが共有しているメモリ内になければならない。前者は主にノード間、後者はノード内並列に用いられる。 - 9.レプリカ交換分子動力学法(REMD法)
タンパク質などの生体高分子の立体構造のダイナミクスや折れ畳みなどをシミュレーションするための効率の良いアルゴリズムの1つ。オリジナルのREMD法では、計算対象に関するコピー(レプリカ)を複数用意し、異なる温度で分子動力学計算を実施し、ある頻度でレプリカ間の温度等を交換することで、低温から高温までの条件が交じった分子動力学シミュレーションを実現する。これによって、特に低温のシミュレーションで起こる初期構造に近い構造に留まる問題などが解決し、多くの立体構造を探索することで、実効的に長時間の分子動力学を行ったのと同じ計算結果を得ることができる。
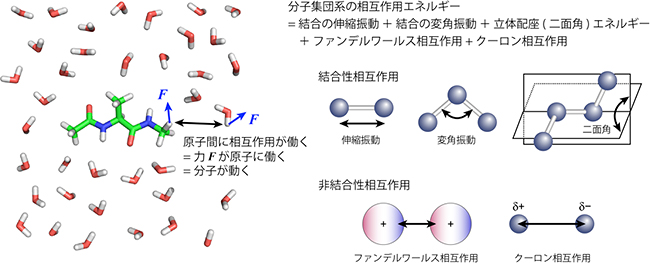
図1 分子集団系の原子間相互作用
分子動力学計算では、原子を「電荷を持った質点」として近似し、それらがバネでつながっている分子モデルを扱う。分子が多数集合した系に対しては、原子同士の相互作用を結合性相互作用(伸縮振動、変角振動、二面角変化に伴う相互作用)と非結合性相互作用(ファンデルワールス相互作用とクーロン相互作用)に分けて計算する。二面角変化に伴う相互作用は、分子中で4個並んだ原子のうち3個から構成される面同士のなす角に関するポテンシャルエネルギーで、シス、トランス、ゴーシュ型などの立体配座を決める。
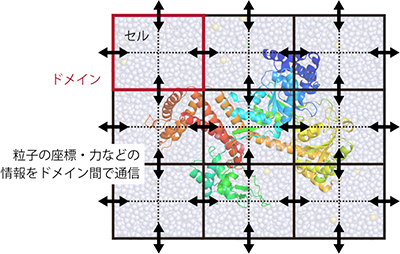
図2 空間分割法の概念図
SPDYNに導入している空間分割法は、シミュレーションの全体空間(ボックス)をドメイン(実線枠)とセル(点線枠)に分割する。MPIプロトコルを用いた並列計算では、各ドメインに各MPIプロセスを割り当て、粒子の座標や力、ドメイン間を移動する粒子の情報などを通信する(矢印)。粒子がボックスから外へ出て行くと、反対側から粒子がボックスの中に入ってくるような周期性を考慮に入れている。
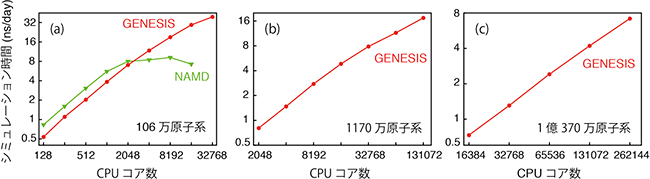
図3 「京」を用いたGENESISのベンチマーク
- (a) サテライトタバコモザイクウイルスのモデル約106万原子系のシミュレーション時間
- (b) バクテリアの細胞質分子混雑環境を模倣した約1,170万原子系のシミュレーション時間
- (c) バクテリアの細胞質分子混雑環境を模倣した約1億370万原子系のシミュレーション時間
約1,170万原子に対して1日あたり17.5ナノ秒(ns)、約1億370万原子系に対して1日あたり6.5nsという性能を「京」コンピュータを用いて達成した。(a)に関しては、米国イリノイ大学の研究グループが開発した世界最高速分子動力学計算ソフトウェアの1つであるNAMDと比較した。
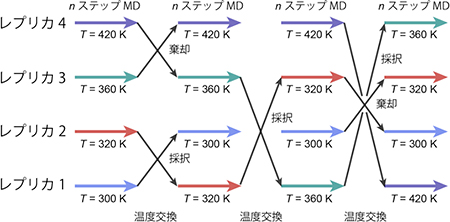
図4 レプリカ交換分子動力学法の概念図
レプリカ交換法では、対象とする系のコピー(レプリカ)を複数用意し、それぞれ異なる温度を割り当てる。それらを並列に計算し、あるステップごとにレプリカ間で温度交換を試みる。交換が採択されれば温度を交換し、棄却されればそのままの温度で計算を進める。