要旨
理化学研究所(理研)情報基盤センター バイオインフォマティクス研究開発ユニットの林哲太郎センター研究員、尾崎遼基礎科学特別研究員、二階堂愛ユニットリーダーらの研究チーム※は、これまで検出が難しかった多様なRNA[1]の発現量と完全長を1細胞で計測できる「1細胞完全長トータルRNAシーケンス法『RamDA-seq』[2]」を開発しました。
細胞の多様性は、ゲノム[1]にコードされた数万の遺伝子[1]領域から転写されるRNAの種類や量によって決まります。そのため、一つ一つの細胞の中に存在するRNAの種類と量が分かれば、どの遺伝子がどのくらい働いているかが分かり、細胞や臓器の状態・機能をより深く理解できます。1細胞に含まれるRNAの種類と量を網羅的に計測する技術は、「1細胞RNAシーケンス法(1細胞RNA-seq[3])」と呼ばれます。最近、非ポリA型RNA[4]が細胞分化や疾患に関与することが明らかになり、大きな注目を集めています。しかし、既存の1細胞RNA-seqでは非ポリA型RNAが検出できないため、非ポリA型RNAが細胞の中で機能していたとしても見逃してしまうという問題がありました。加えて、従来法にはRNAの全長が計測できずに途中で欠損する問題もありました。そのため、ゲノムDNAから転写された全てのRNAについて、ポリA型・非ポリA型を問わず、全長を偏りなく計測するために、新しい技術を開発する必要がありました。
今回、研究チームは、林センター研究員が新たに開発した核酸増幅法RT-RamDA法[5]とランダムプライミング法[6]を組み合わせ、「1細胞完全長トータルRNAシーケンス法『RamDA-seq』」を開発しました。従来法との性能比較の結果、RamDA-seqは非ポリA型RNAを含む約2倍の遺伝子種を精度よく検出でき、どんなに長いRNAでもほぼ全長の配列を計測できることを確認しました。また、マウス胚性幹細胞(ES細胞)[7]を用いた検証の結果、従来法では計測できなかったヒストンmRNA、長鎖ノンコーディングRNA(lncRNA)[8]のNeat1、エンハンサーRNA[9]といった非ポリA型RNAの細胞間での変動を計測できました。さらに、30万塩基を超える非常に長い新生RNA[8]を捉えられました。
本成果は今後、細胞分化や臓器・器官発生などの基礎研究から、再生医療における移植細胞の安全性評価、血中循環腫瘍細胞など希少細胞集団の診断マーカーの開発まで、あらゆるライフサイエンスの研究分野の発展に貢献すると期待できます。
本研究は、英国のオンライン科学雑誌『Nature Communications』(2月12日付け)に掲載されました。
本研究は、日本医療研究開発機構(AMED)創薬等ライフサイエンス研究支援基盤事業(PDIS)、科学技術振興機構(JST)およびAMED 再生医療実現拠点ネットワークプログラム、文部科学省科学研究費、日本学術振興会(JSPS)科学研究費の支援を受けて行われました。また、本研究の一部は、JSTのCREST「臓器・組織内未知細胞の命運・機能の1細胞オミクス同時計測」の支援を受けました。
※研究チーム
理化学研究所 情報基盤センター バイオインフォマティクス研究開発ユニット
センター研究員 林 哲太郎(はやし てつたろう)
基礎科学特別研究員 尾崎 遼 (おざき はるか)
上級センター研究員 笹川 洋平(ささがわ ようへい)
テクニカルスタッフI 梅田 茉奈(うめだ まな)
センター研究員(研究当時) 團野 宏樹(だんの ひろき)
ユニットリーダー 二階堂 愛(にかいどう いとし)(多細胞システム形成研究センター 一細胞オミックス研究ユニット ユニットリーダー)
背景
生命の基本単位である細胞は、同じゲノム配列を持ちながら多様な機能に分化し、私たちの体を構成しています。一つの臓器は複数の細胞種で構成されており、さらに同じ細胞種であっても、一つ一つの細胞に含まれるRNAの種類と量は異なります。このような細胞の多様性は、ゲノムにコードされた数万の遺伝子領域から転写されるRNAが、どのような組み合わせで作られるかによって決まります(図1)。これはRNAそのものや、RNAから翻訳されるタンパク質が、細胞の構造や機能を司るためです。つまり、1細胞ごとにRNAを網羅的に計測することが、その臓器の成り立ちや状態、疾患などを理解する上で非常に重要となります。
これを実現する技術が、「1細胞RNAシーケンス法(1細胞RNA-seq)」です。1細胞RNA-seqでは、RNAをDNAに変換し、ハイスループットDNAシーケンサーによって配列決定することで、一つの細胞に含まれる10ピコグラム(pg、1pgは1兆分の1グラム)という微量のRNAの種類や量を配列情報として網羅的に計測することができます。これまでに、多くの1細胞RNA-seq技術が開発され、発生・幹細胞・がん研究などさまざまな研究現場で活用されてきました。
RNAは、末端にポリA配列を持つポリA型RNAと、ポリA配列を持たない非ポリA型RNAに分けられます(図1)。ポリA型RNAには、タンパク質をコードするメッセンジャーRNA(mRNA)のほとんどが含まれています。一方、非ポリA型RNAには、ヒストンタンパク質をコードするmRNA(ヒストンmRNA)、長鎖ノンコーディングRNA(lncRNA)、新生RNA、環状RNA[8]、エンハンサーRNAなどが含まれており、その多くは機能が分かっていませんでした。しかし近年になり、非ポリA型RNAが細胞分化や疾患、遺伝子発現の制御といった重要な生命現象に関与することが明らかになり、非ポリA型RNAを計測する重要性が高まっています。
また、RNAには数十塩基から数十万塩基までさまざまな長さのものがあります。さらに、スプライシング[10]という過程により、細胞の種類や状態によって同じ遺伝子から長さと配列が異なるRNAが転写されることがあります(図1)。長さと配列が異なるRNAはRNA自体やそこから翻訳されるタンパク質の機能が異なったり、場合によっては疾患の原因となったりすることがあります。このようなRNAを見分けるにはRNAの全長を計測する必要があります。
しかし、既存の1細胞RNA-seqでは、非ポリA型RNAや非常に長いRNAの全長を捉えることができません。原因は1細胞RNA-seqの原理にあります。1細胞RNA-seqでは、RNAをDNAに変換する際に、RNAを鋳型に相補的DNA(cDNA)を合成する逆転写反応[11]を利用します。この逆転写反応の開始には、逆転写プライマーという短いDNA配列が必要で、既存の1細胞RNA-seqではオリゴdTプライマー[12]を用います。オリゴdTプライマーはポリA型RNAと結合しますが、非ポリA型RNAに結合しないため、非ポリA型RNAはcDNAに変換されません。また、オリゴdTプライマーを用いるとRNAの末端からcDNAを合成することになるため、長いRNAや複雑な二次構造を持つRNAは、cDNAの合成が途中で止まってしまい、全長を捉えることができません。さらに、cDNAを増幅させるポリメラーゼ連鎖反応(PCR)[13]は、増幅用共通配列を付加した逆転写プライマーが必須で、かつDNAの長さに依存した増幅バイアスもあるため、感度や再現性が低下することが知られています。
このような技術的制約から、ポリA型・非ポリA型を問わずRNAの全長を偏りなく計測するためには、オリゴdTプライマーにも、PCRによる増幅にも頼らない、全く新しい原理による技術の開発が必要でした。
研究手法と成果
研究チームは、1細胞レベルでポリA型・非ポリA型を問わず、RNAの全長を網羅的に計測できる「1細胞完全長トータルRNAシーケンス法」の開発に取り組みました。具体的には、逆転写反応の際にオリゴdTプライマーではなく、ランダムプライマー[6]を用いるランダムプライミング法を1細胞RNA-seqに応用することを目指しました。そのために、以下の二つの課題を解決しました。
- 1.リボソームRNA(rRNA)[14]の逆転写の抑制
細胞内に存在するRNAの9割以上は計測してもあまり意味のないrRNAです。ランダムプライミング法ではrRNAがcDNAに合成され、他のRNAの情報が失われてしまいます。そこで、rRNAを認識する配列を除いたランダムプライマーである 「not-so-random プライマー(NSRs)」を使用しました。NSRsを用いることで、rRNA由来のcDNA合成を抑えつつ、ポリA型RNAおよび非ポリA型RNAを計測することが可能になりました。
- 2.逆転写効率の向上
ランダムプライマーに増幅用の共通配列を付加すると、逆転写効率が著しく低下します。これでは、存在量の少ないRNAは捉えることができません。そこで、林センター研究員が開発した新しい核酸増幅法「RT-RamDA法」を用いました。RT-RamDA法は、逆転写反応中にRNAから直接cDNAを増幅できる新しい核酸増幅法です。既存の核酸増幅法はcDNAの増幅の前に複数の反応ステップを経る必要があるため、途中で一部のRNAが失われる問題がありました。しかし、RT-RamDA法は、最初の反応ステップでRNAから直接cDNAを増幅できるため、高効率な逆転写と高感度なcDNA増幅が可能となりました。
このようにRT-RamDA法とNSRsを組み合わせることで、1細胞完全長トータルRNAシーケンシング法である「RamDA-seq(ラムダセック)」を確立しました(図2)。RamDA-seqでは逆転写反応のみで増幅が完了するため、従来法で問題となっていたRNAの捉え漏れやPCRによる増幅バイアスを避けられるとともに、操作が簡便になり試薬コストも削減されました。
RamDA-seqの性能を評価するため、1細胞相当量にあたる10 pg のRNAを用いて、既存の1細胞RNA-seqと比較しました。まず、非ポリA型RNAを含む遺伝子の検出数を比較したところ、RamDA-seqは、既存の1細胞RNA-seqと比べて最も検出できる遺伝子数が多いことが分かりました(図3)。従来法との性能比較の結果、RamDA-seqは非ポリA型RNAを含む約2倍の遺伝子種を精度よく検出できました。また、複数回の独立した実験間での再現性も既存技術よりも優れていました。さらに、RamDA-seqは既存の1細胞RNA-seqに比べて、長鎖のRNAであっても全長を偏りなく計測できることが分かりました。例えば、長さが1万塩基以上という非常に長鎖のRNAにおいて、既存の1細胞RNA-seqでは多くのエキソン(遺伝情報がコードされている部分)が計測できていないのに対し、RamDA-seqでは全てのエキソンが計測できました(図4)。これらの結果から、RamDA-seqがポリA型・非ポリA型を問わずRNAの全長を偏りなく計測できる完全長1細胞トータルRNA-seqであることが確認できました。
続いて、RamDA-seqを用いて、細胞状態の変化に伴う非ポリA型RNAの発現変動の検出を試みました。マウス胚性幹細胞(ES細胞)を細胞分化誘導後に複数の時刻にサンプリングした細胞に対してRamDA-seqを適用し解析した結果、これまでに知られていない新しいRNAを含む458種類の非ポリA型RNAが、細胞分化の進行に伴ってダイナミックに変動していることが分かりました。なかでも、腫瘍抑制などさまざまな機能を持ち、2万塩基以上の長さの非ポリA型lncRNA Neat1の変動を捉えることに成功しました。
次に、RNA生合成の重要な段階であるスプライシングの過程をRamDA-seqで捉えられるかを調べました。スプライシングにおいて転写されたRNA分子からイントロン領域(遺伝情報がコードされていない部分)が切り出されます。スプライシングが完了していない新生RNAは、イントロン領域を含む非ポリA型RNAです。実際、RamDA-seqではイントロン領域由来のRNAが多く計測されており、新生RNAが計測できることが分かりました。さらに、イントロン領域の計測値の分布の形から、多段階スプライシング[10]という特殊なスプライシング機構を1細胞レベルで初めて検出することができました(図5)。
さらに、RamDA-seqでエンハンサーRNAを計測できるかを検証しました。エンハンサーRNAはエンハンサー(遺伝子領域から離れて位置し、遺伝子の転写効率を調節する領域)から転写されるRNAで、その多くが非ポリA型RNAです。エンハンサーRNAはエンハンサーの活性を反映し、また、転写制御に直接関わる例も知られています。多くのエンハンサーRNAは遺伝子アノテーション[15]に含まれていないことから、エンハンサーRNAのカタログを用意して解析しました。その結果、RamDA-seqでエンハンサーRNAが計測できることを確認しました。これは、初めて1細胞RNA-seqでエンハンサーRNAを計測した報告です。さらに、細胞分化の進行に伴って変動する1,338種類のエンハンサーRNAを発見することができました(図6)。
今後の期待
近年、lncRNAやエンハンサーRNAを含む非ポリA型RNAが、細胞分化や疾患に関与することが報告されています。また、RNAのスプライシングは一つの遺伝子から多様なRNAおよびタンパク質を作り出す重要な仕組みであり、スプライシングの異常はがんなど疾患の原因になります(図1)。今回開発したRamDA-seqによって、これらのRNAを1細胞ごとに網羅的に計測できるようなりました。研究チームは、すでにほかの研究機関とRamDA-seqを活用した共同研究を実施しています。その過程で、多様な細胞種・生物種の約1万個の1細胞のシーケンスに成功しています。これらの成果は、今後、細胞分化や臓器・器官発生などの基礎研究から、再生医療における移植細胞の安全性評価、血中循環腫瘍細胞など希少細胞集団の診断マーカーの開発まで、あらゆるライフサイエンスの研究分野の発展に貢献すると期待できます。
現在、ヒトの全細胞種類について網羅的、かつ、1細胞レベルでRNAを計測しデータベース化する国際プロジェクトHuman Cell Atlasが、米国・欧州を中心に開始しています。しかし、このプロジェクトで利用される1細胞RNAシーケンス法は、非ポリA型RNAやRNA完全長配列を捉えられません。RamDA-seq法は、Human Cell Atlasを補完するデータベース作成に貢献することが期待されます。このようなデータベースは、核酸医薬の開発に貢献します。核酸医薬では、対象とする細胞に発現するポリA型・非ポリA型RNAの種類と全長配列の情報が、医薬品の効果や副作用の評価に必須となります。そのため、RamDA-seqは、創薬研究にも貢献すると考えられます。
原論文情報
- Tetsutaro Hayashi, Haruka Ozaki, Yohei Sasagawa, Mana Umeda, Hiroki Danno and Itoshi Nikaido, "Full-length total RNA sequencing from single cells uncovers dynamics of recursive splicing and enhancer RNAs", Nature Communications, doi: 10.1038/s41467-018-02866-0
発表者
理化学研究所
情報基盤センター バイオインフォマティクス研究開発ユニット
センター研究員 林 哲太郎 (はやし てつたろう)
基礎科学特別研究員 尾崎 遼 (おざき はるか)
ユニットリーダー 二階堂 愛 (にかいどう いとし)
(多細胞システム形成研究センター 一細胞オミックス研究ユニット ユニットリーダー)
 林 哲太郎
林 哲太郎
 尾崎 遼
尾崎 遼
 二階堂 愛
二階堂 愛
報道担当
理化学研究所 広報室 報道担当
Tel: 048-467-9272 / Fax: 048-462-4715
お問い合わせフォーム
産業利用に関するお問い合わせ
理化学研究所 産業連携本部 連携推進部
お問い合わせフォーム
AMED事業に関するお問い合わせ
日本医療研究開発機構(AMED)
創薬戦略部 医薬品研究課
Tel: 03-6870-2219 / Fax: 03-6870-2244
20-DDLSG-16 [at] amed.go.jp(※[at]は@に置き換えてください。)
戦略推進部 再生医療研究課
Tel: 03-6870-2220 / Fax: 03-6870-2242
saisei [at] amed.go.jp(※[at]は@に置き換えてください。)
補足説明
- 1.RNA、ゲノム、遺伝子
ゲノムは細胞が持つ全遺伝情報であり、DNA(デオキシリボ核酸)という生体高分子に4種類の塩基の配列として記録されている。ゲノムの中の遺伝情報が記録(コード)された領域が遺伝子である。遺伝子領域の情報は、DNAを鋳型としてRNA(リボ核酸)が合成(転写)されることで読み出される。 - 2.1細胞完全長トータルRNAシーケンス法『RamDA-seq』
多様なRNAの発現量と全長を1細胞で計測する方法。rRNAを除いたRNAを用いることで、rRNA以外の非ポリA型及びポリA型の全てのRNAとその全長を検出することができるRNAシーケンス法。ヒストンmRNA、長鎖ノンコーディングRNA、新生RNA、環状RNA、エンハンサーRNAなどを検出できる。大量のRNAを用いたトータルRNAシーケンスでは、最初にrRNAを除去するステップがあるが、その際、RNAのロスが生じる。また操作が煩雑となるため、1細胞に適応することは難しい。RamDA-seq(ラムダセック) は、Random Displacement Amplification sequencingの略。 - 3.1細胞RNA-seq
1細胞中に含まれるRNAを、ハイスループットDNAシーケンサーを用いて配列決定し、網羅的かつ定量的にその量や種類を決定する方法。微量なRNAを用いるため、微量RNAからの逆転写反応とシーケンス可能な量までcDNAを増幅させる二つのステップが必要である。 - 4.非ポリA型RNA
タンパク質をコードするmRNAと異なり、RNAの3’末端にポリA配列(アデニン塩基が連続してつながったもの)を持たないRNAの総称。ヒストンタンパク質をコードするヒストンmRNAは例外的にポリA配列を持たない。 - 5.RT-RamDA法
逆転写反応中にRNAから直接、全遺伝子増幅を可能にした新しい核酸増幅法(特許申請中)。DNAを鋳型とする従来の増幅法と異なり、増幅用の共通配列を必要としない。このため、逆転写プライマーに増幅用配列を付加する必要がなく高効率な逆転写が可能となる。また、少ないステップで増幅が完了するため、従来法と比べて収率が高く高感度である。RT-RamDAは、Reverse Transcription with Random Displacement Amplification の略。 - 6.ランダムプライミング法、ランダムプライマー
ランダムプライマーは逆転写プライマーの一つ。アデニン、チミン、シトシン、グアニンの4種類の塩基がランダムに6塩基がつながったDNA鎖。RNAの配列に関係なく結合できるためポリA型RNAと非ポリA型RNA両方に結合することができる。また、RNAの末端だけでなく、RNAの全面に結合することができる。ランダムプライミング法はランダムプライマーを用いた逆転写反応で、RNAの捉え漏れの低減やRNAの全長をcDNAに変換できるメリットがある。 - 7.胚性幹細胞(ES細胞)
ほ乳類の着床前胚(胚盤胞)に存在する内部細胞塊から作製した細胞株で、体を構成する全ての種類の細胞に分化する能力(多能性)を持つもの。 - 8.長鎖ノンコーディングRNA(lncRNA)、新生RNA、環状RNA
lncRNAはタンパク質をコードしない300塩基以上のRNA。非ポリA型RNAが多く含まれ、さまざまな機能を持つことが報告されている。新生RNAはスプライシングが完了していないRNAで、通常非ポリA型RNAである。環状RNAは、端が存在しない環状のRNAで、非ポリA型RNAである。 - 9.エンハンサーRNA
エンハンサーから転写されるRNA。エンハンサー自体の活性を反映し、転写制御に直接関わる例も知られている。 - 10.スプライシング、多段階スプライシング
通常のスプライシングでは、イントロンの上流側の端にある5’スプライス部位から下流側の端にある3’スプライス部位までの領域が切り出される。一方、多段階スプライシングの場合、イントロンの途中に3’スプライス部位と5’スプライス部位が隣接して存在する部位(RS部位)があり、一度のスプライシングでRS部位の3’スプライス部位までが切り出され、続いてRS部位の5’スプライス部位から次のスプライシングで切り出される。 - 11.逆転写反応
RNAを鋳型とし相補的なDNA(cDNA)を合成する反応。RNAウイルスが宿主細胞に進入し自己複製する際に働くRNA依存性DNA合成酵素である逆転写酵素を利用する。逆転写酵素がcDNA合成を行うには、RNA配列に相補的に結合した短いDNA鎖(逆転写プライマー)を出発点とする必要がある。 - 12.オリゴdTプライマー
逆転写プライマーの一つ。ポリA型RNAの末端に付加されるポリA配列に結合するチミン塩基が20個程度連続的につながったDNA鎖。非ポリA型RNAには結合できない。 - 13.ポリメラーゼ連鎖反応(PCR)
熱耐性を持つDNA依存性DNA合成酵素を用いて、鋳型DNAとPCRプライマーの結合、相補DNAの合成、二本鎖DNAの解離の3ステップを繰り返すことで、DNAを連続的かつ指数関数的に増幅させることができる反応。鋳型DNAの長さや配列に依存した増幅バイアスが生じる。長いDNAは増えにくい。さまざまな種類のRNAを由来とするcDNAを増幅させるには、PCRプライマーが結合するための共通配列を予め逆転写プライマーに付加させておく必要がある。PCRはPolymerase Chain Reactionの略。 - 14.リボソームRNA(rRNA)
タンパク質が翻訳される過程で機能する非ポリA型RNA。細胞中に全RNAの90%以上がrRNAといわれている。シーケンスをしてもあまり意味のないRNA種。 - 15.遺伝子アノテーション
ここでは特に、遺伝子がゲノム上のどこに存在し、遺伝子からどのようなRNAが転写されるかをまとめたカタログを指す。これまで多くのRNAが登録されてきたが、細胞内に存在する全てのRNAが登録されるには至っていない。
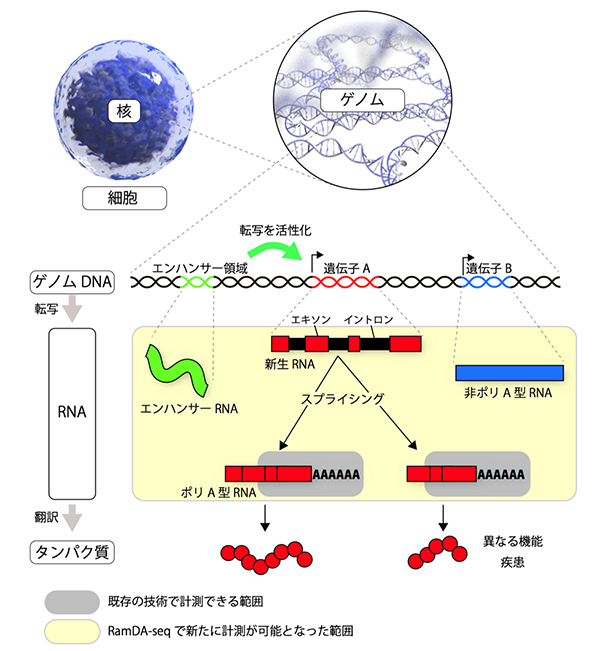
図1 ゲノムから遺伝子、RNA、タンパク質まで
我々の身体を形成する全ての細胞は、共通の遺伝情報を持つ。この遺伝情報をゲノムといい、細胞核内にあるDNAにその情報は保存されている(ゲノムDNA)。ゲノムDNA上には遺伝情報が記録されている遺伝子領域とそうでない領域があり、遺伝子領域からはRNAが転写される。転写された直後のRNA(新生RNA)にも遺伝情報を持つ領域(エキソン)と持たない領域(イントロン)が存在するため、スプライシングという機構でイントロンが取り除かれ成熟したRNAとなる。成熟RNAは、末端にアデニン(A)が連続的に付加されてポリA型RNAになるものと、Aが付かない非ポリA型RNAになるものに分類される。ポリA型RNAとなる遺伝子の多くは、タンパク質に翻訳されて機能するが、非ポリA型RNAとなる遺伝子は、一部の例外を除いてタンパク質に翻訳されずRNAのままで機能する。スプライシングが変化すると、RNAの長さと配列が変わり、翻訳されるタンパク質の機能が変わったり、疾患の原因となる。また、遺伝子領域外には、遺伝子の転写活性を調整するエンハンサー領域があり、最近になって、エンハンサー領域から転写されるRNA(エンハンサーRNA)が発見された。
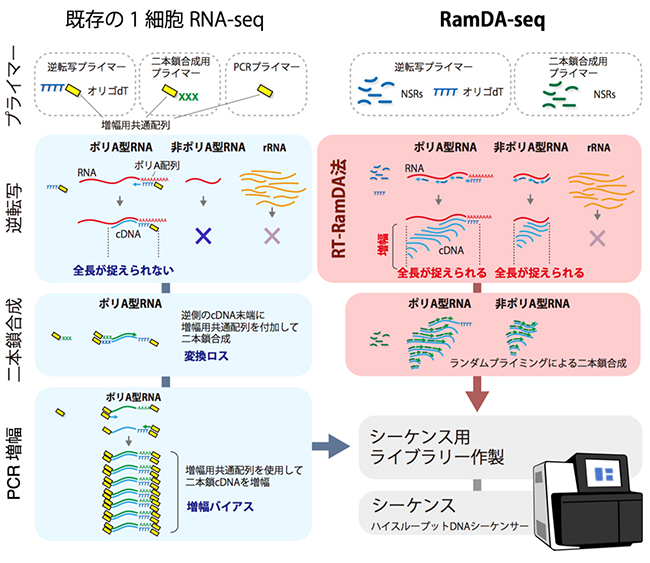
図2 既存の1細胞RNA-seqとRamDA-seqのワークフロー
- 左) 既存の1細胞RNA-seqは、オリゴdTプライマーのみを用いて逆転写を行うため、ポリA型RNAしかcDNA合成ができない。また、長いRNAは、cDNA合成が端までたどり着く前に止まるため、全長を捉えることができない。さらに、分子の数が少ない状態でcDNAを二本鎖合成するため、合成に失敗した分子は確実に失われる(変換ロス)。
- 右) RamDA-seqは、not-so-random プライマー(NSRs)を用いたランダムプライミングで逆転写を行うため、非ポリA型RNAも捉えることができる。また、RNAの全面にプライマーが結合できるため、どんなに長いRNAであっても全長を捉えることができる。加えて、RT-RamDAでは逆転写をしながらcDNAの増幅が行われ、分子の数が多くなるため、二本鎖合成のステップでの変換ロスの影響を受けにくい。さらにPCRが不要なため、DNAの長さや配列による増幅バイアスの低減や操作の簡略化、試薬コストの削減につながった。
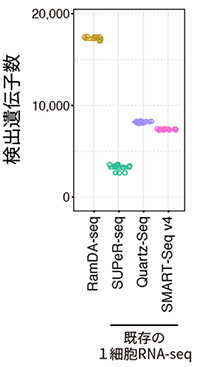
図3 検出遺伝子数の比較
陽性対照であるトータルRNAシーケンシング法を発現遺伝子の正解とし、検出できた遺伝子(トランスクリプト)の数を比較した。RamDA-seqは既存技術に比べ、多くの種類のRNAを計測できることが分かる。
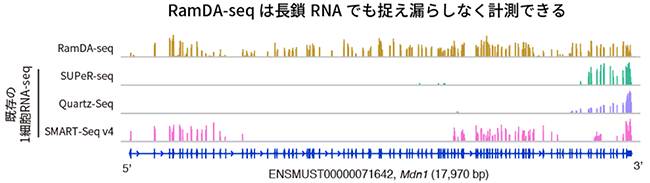
図4 長鎖RNAにおけるRNA計測の比較
102個のエキソン(遺伝情報がコードされている部分)を持つ長さ17,970塩基の長鎖RNAであるMdn1について、全長にわたって計測できているかを調べた。既存技術では多くのエキソンが計測できていないのに対し、RamDA-seqでは全てのエキソンが計測できていることが分かる。
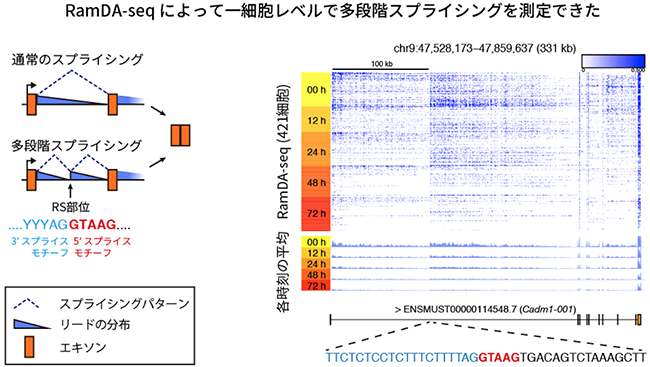
図5 特殊なスプライシングの検出
- 左) スプライシングの種類と計測値(リード)の分布の関係。計測値の分布のパターンはスプライシングの種類を反映する。多段階スプライシングの場合、イントロンの中に矩形波様のパターンが観察される。
- 右) Cadm1遺伝子の第1イントロンにおけるRamDA-seqのリードの分布。分化誘導後の各時刻にサンプリングしたマウスES細胞に対してRamDA-seqを適用した。多段階スプライシングを示すリードの分布(青)が観察された。
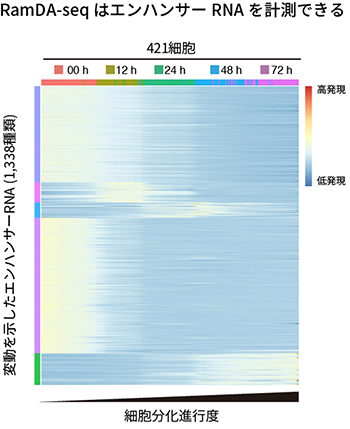
図6 細胞分化の進行に伴って変動するエンハンサーRNAの検出
理研が主宰する国際研究コンソーシアム「FANTOM5」プロジェクトによって報告されたエンハンサーRNAをRamDA-seqで検出した。変動を示すエンハンサーRNAが1,338種類発見できた。横軸は細胞分化の進行度に従って並べ替えた細胞、縦軸はエンハンサーRNA(色は類似した発現パターンを共有するクラスターを表す)、各セルの色はエンハンサーRNAの発現量(正規化されている)を表す。