2010年10月25日
独立行政法人 理化学研究所
次世代シークエンサーで、日本人の全ゲノム配列を包括的解析
-日本人の塩基配列の多様性を詳細に解析した初報告-
ポイント
- 一塩基多様性、コピー数の多様性、構造の多様性を正確に調べる方法を提案
- ヒトゲノム計画による参照配列にない新規配列3百万塩基対を同定・解析
- 全ゲノム配列の高精度な解析で、病気にかかわる未発掘の多様性の解明に期待
要旨
独立行政法人理化学研究所(野依良治理事長)は、次世代シークエンサー(DNA解析装置)※1を用いて日本人1人の全ゲノムのDNA塩基配列(シークエンス)データを初めて包括的に解析しました。理研ゲノム医科学研究センター(鎌谷直之センター長)情報解析研究チームの藤本明洋特別研究員、角田達彦チームリーダー、バイオマーカー探索・開発チームの中川英刀チームリーダーらによる成果です。
現在、塩基配列の違いにより発症リスクが異なる病気の解明に向けて、ゲノムワイド関連解析※2が爆発的に行われていますが、それに続く次世代の方法として、ヒトの全ゲノム配列(約30億塩基対)を解析する方法「全ゲノムシークエンス解析」が期待されています。しかし、いまだに精度良く解析する方法が確立していない上、日本人についてはまだ報告がありませんでした。研究グループは今回、次世代シークエンサーを使って、日本人男性1人の全ゲノム配列の高精度な解析を達成しました。取得した全ゲノムシークエンスデータの99%以上は、ヒトゲノムプロジェクト※3で決定された参照配列にマップできました。このデータにベイズ決定法※4という数学的手法を適用し、約313万個の一塩基多様性※5を約99.9%の高精度で検出しました。そして、海外の別々の研究グループから報告されている欧米人、アフリカ人、中国人、韓国人の6人の全ゲノム配列と日本人の全ゲノム配列を比較し、集団では見失われていた、遺伝子の機能に影響を与える一塩基多様性が個人個人には多いことを発見しました。また、高精度な方法で1万塩基対より小さい欠失を約5,300個検出し、コピー数の多様性※5や構造の多様性※5も網羅的に見いだしました。さらに、ヒトゲノム参照配列にない約300万塩基対の新規配列を発見し、これらの配列がヒトゲノムの多様性を反映する可能性を見いだしました。一連の解析で、ヒトゲノムには未発掘の多様性に富んだDNA塩基配列が数多く存在し、全ゲノムシークエンス解析は、それらを完全に理解するために極めて重要なアプローチであることが分かりました。今後、このような方法で日本人固有の多様性を検出することによって、日本人のための病気の研究への展開が期待できます。
本研究成果は、米国の科学雑誌『Nature Genetics』オンライン版(10月24日付け:日本時間10月25日)に掲載されます。
背景
理研ゲノム医科学研究センターは、2002年から世界に先駆けてゲノムワイド関連解析という方法を確立し、さまざまな病気にかかわる遺伝子を明らかにしてきました。また、国際ハップマッププロジェクト※6に参画し、その成果を基に、病気にかかわる遺伝子を探索するための一塩基多型(SNP)の効率的なセットも整備しました。このような遺伝子多型解析技術の進展によって、ゲノムワイド関連解析を用いた病気にかかわる遺伝子の解明などが、今や全世界で劇的な展開を見せています。しかし、この方法は、集団内で多くの人が持つ「多型」に着目したもので、より頻度の低い「多様性」まではカバーしきれていません。頻度の低い多様性を探索する現時点で唯一の方法は、ヒトの全ゲノム配列(約30億塩基対)を解析する方法「全ゲノムシークエンス解析」です。特に、この数年で爆発的に解析能力が向上してきている次世代シークエンサー(DNA解析装置)を活用した超並列シークエンス技術が最も強力で、余すことなくDNA塩基配列を解析できる方法となっており、将来のオーダーメイド医療にも有用であると期待されています。次世代シークエンサーによる全ゲノムシークエンス解析を行った海外の先行研究から、一塩基多様性、コピー数の多様性、挿入/欠失、転座などの構造の多様性に関する情報が豊富に得られることが分かってきました。しかし、全ゲノムシークエンス解析による多様性検出の精度は、実験手法固有のエラー、マッピングエラー、ヒトゲノムプロジェクトで決定された参照ゲノム配列との集団としての違い、検出アルゴリズムの違いなどによって影響を受け、次世代シークエンサーを活用した高精度な方法はいまだ確立しているとはいえません。これらの課題を克服するためには、シークエンス技術の改良とともに、より洗練された情報科学的アプローチが必要となっています。また、日本人の全ゲノム配列の包括的解析も進んでおらず、固有の配列や多様性があるのか無いのか、どのような描像なのかなども、不明なままでした。
研究手法と成果
研究グループは、米国・イルミナ社のGenome Analyzer IIという次世代シークエンサーを活用し、国際ハップマッププロジェクトで解析された日本人男性1人のDNAから、全部で約1,200億塩基対(精度を良くするために各塩基対につき約40回分)のデータを得ました。その99%以上が、米国立生物工学情報センター(NCBI)に登録されているヒトゲノム参照配列(約30億塩基対)にマップすることができました。
一塩基多様性の検出のため、いくつかの数学的手法を比較し、最終的に成績の良かったベイズ決定法を用いることにしました。ベイズ決定法によって検出した一塩基多様性のうち、既知の一塩基多型(SNP)と重なるものは、その約99.9%で遺伝子型が合致し、この方法が高精度であることを確かめました。この方法で見つけた3,132,608個のSNPのうち、12.6%の395,940個は既知のデータベースに無く、新規のものでした。また、タンパク質コード領域内で、アミノ酸配列が異なる9,783個の塩基の多様性と、遺伝子機能を喪失する96個の塩基の多様性を見つけました。さらに、217,176個の短い配列の挿入や228,063個の短い配列の欠失を検出しましたが、そのうち487個はタンパク質コード領域内に存在していました。タンパク質コード領域内には、アミノ酸配列を途中から崩す(3文字単位でない)塩基対の挿入/欠失も351個見つかりました。これらの多様性はいずれも、遺伝子の機能に影響を与えている可能性があります。
日本人1人の全ゲノム配列と、海外の複数のグループの先行研究から得られている欧米人、アフリカ人、中国人、韓国人の6人の全ゲノム配列の一塩基多様性のデータを合わせて解析したところ、個人個人には、集団では見失われていたアミノ酸配列が異なる塩基の多様性や遺伝子機能を喪失する塩基の多様性が多いことが分かりました(図1)。この結果から、遺伝子機能に良くない影響を与える一塩基多様性のほとんどが、自然選択のためにまれになるため、これまでの集団内での一塩基多型(SNP)の探索では、大多数のものが見失われてきたことが推測できます。また、遺伝子機能別に分類して解析してみると、遺伝子機能を喪失する塩基の違いは、嗅覚や化学的刺激の認識にかかわるものに多いことが分かりました。
配列の欠失の検出には、各塩基対が読まれた回数(リードの深さ)と、リード対間の距離の両方を情報として用いる高精度な方法を実現しました(図2)。その結果、5,319個の欠失の候補が挙がりました。それらの一部をポリメラーゼ連鎖反応(PCR)法で検証すると、すべて欠失であることが分かりました。この方法を用いると、これまでのアレイ技術では検出が難しい、数百塩基対の小さな欠失を検出することができます。検出した欠失のうち74個が、70個の遺伝子領域(126個のエキソン)と重なることが分かりました。このような欠失は遺伝子の機能に影響を与えている可能性があります。
1万塩基対以上という長い配列のコピー数多様性の検出には、5,000塩基対の範囲内の読まれた回数を用いることにしました。その結果、コピー数が多い領域113個と、コピー数が少ない領域109個を検出しました。それらを別の実験で検証したところ、結果が良く一致することが分かりました。この技術の大きな特徴は、ほかのサンプルと比較することなく、1サンプルだけで検出が可能なことです。また57個の染色体上で配列が逆転する逆位や112個の一部がほかの場所と入れ替わる染色体内転座の候補も見いだしました。
塩基配列の組み立てを行うABySS、SOAPdenovo、Velvetという3種類のソフトウエアを用いて、ヒトゲノム参照配列にマップできなかったデータを組み立てた(アセンブル)結果、それぞれ6,535個、4,826個、6,617個の連続した配列断片(コンティグ)を得ました。この配列断片は新規配列にあたり、3つのソフトウエアが出す結果は、互いによく似ていました。そして配列断片の185個をポリメラーゼ連鎖反応(PCR)法で検証したところ、181個が実際に存在することが分かり、さらに、新規配列の90%以上が、相当する領域を通常のシークエンス解析で決定する検証実験でも、同じ配列が得られることを確認しました。今回の全ゲノムシークエンス解析では、全部で300万から340万の塩基対がヒトゲノム参照配列に無い新規の配列で、ヒトゲノムの多様性を反映するものと考えられます。
今後の期待
一般に、有害な遺伝的多様性は、自然選択のために集団内では抑えられていると考えられますが、個人のゲノム配列上には、病気にかかわるまれな多様性が未発掘のままとなっている可能性があります。全ゲノムシークエンス解析は、そのようなまれな多様性を余すことなく検出する本質的な技術となりえます。全ゲノムシークエンス解析のもう1つの特徴は、ヒトゲノム参照配列にない新規配列を発見できることです。2003年に配列決定が完了したとされているヒトゲノム配列には、未発見の配列や多様性が多く存在すると考えられ、今後数年から5、6年ぐらいの間に、全ゲノムシークエンス解析によってさまざまな病気にかかわる未知の多様性が発見されると予想されます。その結果、オーダーメイド医療がますます進展し、病気の研究に新たな展開をもたらすことが期待されます。また、今回の解析技術を駆使することによって、ゲノムの病気であるがんのゲノム解析を行うICGC (国際がんゲノムコンソーシアム)が進捗し、がんにかかわるゲノム上の包括的情報を解明していくと注目されます。
発表者
理化学研究所
ゲノム医科学研究センター 情報解析研究チーム
チームリーダー 角田 達彦(つのだ たつひこ)
Tel: 045-503-9556 / Fax: 045-503-9555
お問い合わせ先
横浜研究推進部 企画課Tel: 045-503-9117 / Fax: 045-503-9113
報道担当
理化学研究所 広報室 報道担当Tel: 048-467-9272 / Fax: 048-462-4715
補足説明
- 1.次世代シークエンサー(DNA解析装置)
現在使われているいわゆる次世代シークエンサーの基本原理は、解析する対象とするDNAを細かく断片化し、それらを超並列に(同時に数千万個所を)解読することである。このため(超)並列シークエンサーとも呼ばれる。断片化したものの端から配列決定したものをリードという。ポリメラーゼの能力に限界があるため、次世代シークエンサーのリードは一般に短いが(本研究では50塩基のリードが多い)、改良とともに、だんだん長く読めるようになっている。断片化したDNAの両端から読んだリードの対をペアエンドリードという。 - 2.ゲノムワイド関連解析
病気に罹患している集団と一般対照集団との間で遺伝情報(アレルの出現頻度など)の違いを検定し、病気の原因となる遺伝子や多型を見いだすことを、全ゲノム領域の各多型に対し行う方法。 - 3.ヒトゲノムプロジェクト
4つの塩基(A, T, C, G)から成る約30億塩基対のヒトの全ゲノム配列を、国際協力で解読するプロジェクトで、1990年に発足し2003年に完了した。解読されたゲノムは、米国立生物工学情報センター(NCBI)などの研究機関で参照することができ、そこにはヒトの全遺伝子の99%の配列が99.99%の正確さで含まれるとされている。ヒトゲノム計画のもう1つの目標は、より高速で効率的なDNAシークエンス法を開発して、それを産業化に向けて技術移転することであり、その後、DNAシークエンス技術の革新的発展が起こり、今日、効率の良い個人の全ゲノムシークエンス解析が可能になりつつある。 - 4.ベイズ決定法
物事を決定するときに用いる数学的考え方の1つ。本研究では、シークエンサーからのデータが1塩基対につき平均して40回分得られる。その中に異なる塩基が同時に観測された場合に、それが単なるシークエンスエラーなのか、あるいは本当に多様性を反映しているのかを、それぞれの場合で確率を求め、それら確率の高低を比較することによって決定する。 - 5.一塩基多様性、コピー数の多様性、構造の多様性
多様性は、集団内の頻度にかかわらず、個人間の違いを総称したものを指す。一般に多型は集団内の頻度が1%以上の個人間の違いを指し、多様性は多型を包含する。この中で、一塩基多様性は、DNA上の一塩基の個人ごとの違いである。コピー数の多様性は、個人ごとに、1細胞あたりの遺伝子のコピー数が違うゲノム配列(領域)のことをいう。通常、ヒトの遺伝子配列は、父方と母方由来の染色体それぞれに1個ずつあり、合わせて2個(2コピー)存在する。それに対し、個人によっては合わせて1コピーしかなかったり(欠失が起こっている)、3コピーあったり(重複や挿入が起こっている)する。このような遺伝子配列の数の個人差をコピー数の多様性という。構造の多様性は、挿入や欠失、染色体上で配列が逆転する逆位、染色体の一部がほかの場所と入れ替わる染色体内転座による多様性の総称である。 - 6.国際ハップマッププロジェクト
ヒトゲノム上の多型情報を臨床応用していくために不可欠なハプロタイプ地図を作成する国際計画。2002年10月に開催した「国際ハップマッププロジェクト戦略会合」で、日・米・英・中・加の協力によりこの地図の作成に取り組むことが合意された。わが国からは、理研遺伝子多型センターの中村祐輔センター長(当時)が研究代表者としてプロジェクトに参加した。同プロジェクトでは、アジア人(日本人を含む)、欧米人、アフリカ人のそれぞれの人種について、各国より血液サンプルを収集するとともに、それらのDNAの多様性を調べ、ハプロタイプ地図の作成を実施することを目標とし、達成された。プロジェクトで得られたデータを基にした、全ゲノム規模の関連解析(ゲノムワイド関連解析)用実験プラットフォームが開発され、現在、多くの関連解析により疾患関連遺伝子が発見されつつある。
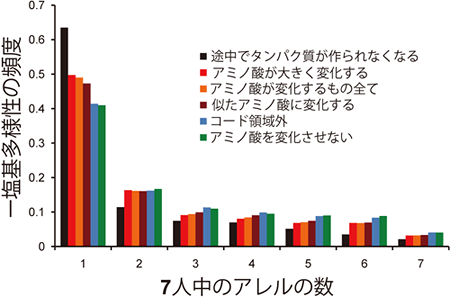
図1 7人(今回の1人と先行研究6人)のゲノムでの一塩基多様性のアレル数の分布
7人の染色体上で、1人の1本の染色体にしか見られない多様性が多い。特に、遺伝子機能を喪失する塩基の多様性や、アミノ酸が異なる塩基の多様性が、個人ごとに調べると多く見つかる。
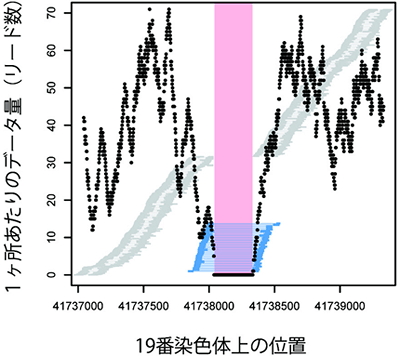
図2 欠失を検出するためのデータの実例
次世代シークエンサーから出力されるデータ(リード)をヒトゲノム参照配列にマップしたとき、データ量(=リード数、黒点の縦軸の位置)が少なく、かつ、本当は近いはずのリードの対(青線の両端)が遠くに引き離されてマップされてしまう場所(ピンク色)は、今回のゲノムで欠失の領域であることを示す。