理化学研究所(理研)生命医科学研究センターがんゲノム研究チームの中川英刀チームリーダー、東京大学医科学研究所健康医療データサイエンス分野の井元清哉教授らを含む国際共同研究グループは、38種類のがんについて、2,800例以上の全ゲノムシーケンス解析[1]を行った結果、4,600万個を超える変異・異常を同定し、その特徴を明らかにしました。本研究は、国際的連携による全ゲノムがん種横断的解析プロジェクト(PCAWG)[2]の一環として行われ、これまでで最も網羅的かつ詳細ながんゲノム情報の解析となりました。
本研究によるゲノム解析基盤の手法とデータ・結果は、次世代のがんゲノム医療や研究における解析基盤の構築に貢献すると期待できます。
今回、PCAWGでは、38種類、2,800例以上に及ぶがんの全ゲノム塩基配列(シーケンス)情報を収集しました。東京大学医科学研究所のスーパーコンピュータ「SHIROKANE」を含む世界10カ所のデータセンターを連結し、標準化された高精度のゲノム解析手法を構築した上で、がん全ゲノムシーケンスの大規模解析を行いました。その結果、非コード領域[3]の変異、大きなゲノム構造異常[4]、ミトコンドリアゲノム[5]異常など、合計4,600万個以上の変異・異常を同定、それらの特徴を明らかにすることで、これまでで最も網羅的かつ詳細ながんゲノムマップ[6]を作成しました。
本研究は、科学雑誌『Nature』(特集号)の掲載に先立ち、オンライン版(2月6日付:日本時間2月6日)に掲載されます。
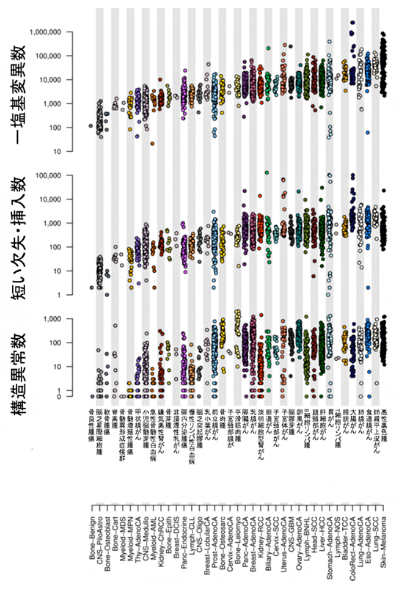
本研究で同定した全ゲノム上での各種がんの変異・異常数
背景
がんは、ゲノムの変異や異常が蓄積することで発症・進行する「ゲノムの病気」です。現在、世界中でがんの網羅的なゲノム解析やゲノム情報に基づく薬の開発・個別化医療(がんゲノム医療)が精力的に行われています。2008年、がんゲノム変異の全容解明とカタログ化を目指し、世界最大規模のがんゲノム国際共同体「国際がんゲノムコンソーシアム(ICGC)[7]」が発足しました。ICGCでは、2018年までに約25,000検体のがんゲノム情報を収集・公開し、理研においても300例の肝臓がんの全ゲノムシーケンス解析を行い、そのデータをICGCで公開しています注1)。
ICGC/TCGA[8]を含むこれまでの主ながんゲノム研究では、全ゲノムの約1~2%に相当するタンパク質をコードする領域(エクソーム)での解析が主になされ、そのデータが蓄積されてきました。しかし、近年の次世代シーケンサー[9]技術の急速な進展、情報解析技術やITハード面の技術革新に伴い、ヒトの約30億塩基対の情報からなる全ゲノムでのシーケンス解析が、容易かつ安価に行えるようになりました。このような背景から、今後は、全ゲノムシーケンス解析が研究のみならず、がんゲノム医療といった診断や個別化医療においても、重要な解析手法になると予測されています。
がん全ゲノムシーケンス解析では、ゲノム解析アルゴリズム(パイプライン[10])によって変異の有無など解析結果が大きく異なる場合があります。そのため、巨大なデータを扱うことができる計算機基盤の構築とともに、解析手法の標準化が求められます。理研を含むICGCの研究グループは、2015年にさまざまなゲノム解析アルゴリズムを比較し、その標準化についてのガイドラインおよびベンチマークを作成しました注2)。
今回、国際共同研究グループは、国際的連携による「全ゲノムがん種横断的解析プロジェクト(PCAWG)」に参加し、がんの大規模な全ゲノムシーケンス解析に取り組みました。本プロジェクトには、37カ国の1,300人以上の科学者、ITエンジニア、臨床家が参加しました。
- 注1)2016年4月12日プレスリリース「肝臓がん300例の全ゲノムを解読」
- 注2)2015年12月9日プレスリリース「がんの全ゲノムシーケンス解析の新たなガイドラインを作成」
研究手法と成果
本研究は2014年に開始し、その時点までにICGC/TCGAで解析された38種類のがんについて2,834例の全ゲノム塩基配列(シーケンス)データを収集しました。そのうちの270例(約10%)は、理研で解析した日本人肝臓がんのデータです。合計約1ペタバイト(1,000兆バイト)に及ぶシーケンス生データを国際連携で効率良く解析するため、大規模な仮想データセンター、高精度・標準化パイプラインというゲノム解析基盤を構築し、変異・ゲノム異常の特徴を明らかにするためのワーキンググループを結成しました(図1)。
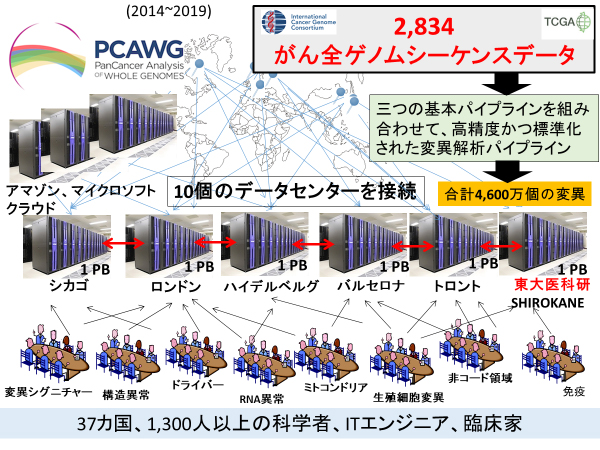
図1 PCAWGでのデータ解析基盤の構築
2,834例のがん全ゲノムシーケンス解析の生データを10個のデータセンターとクラウドで共有し、三つの基本パイプラインを組み合わせた、高精度で標準化された変異同定アルゴリズムを用いて解析した。同定された合計4,600万個の変異情報を下段で示したような16のワーキンググループで共有し、個々の特徴を明らかにした。
- (1)大規模仮想データセンターの構築
世界10カ所にあるゲノム解析センターのスーパーコンピュータをデータセンターとし、アマゾンやマイクロソフトのクラウドサーバーとも仮想マシーン(VM)[11]で接続することでこれらを同期させ、統一された計算空間を構築しました。データセンターとして、日本からは東京大学医科学研究所のスーパーコンピュータ「SHIROKANE」が参加しました。研究に参加する世界中の研究者は、これらのデータに容易にアクセスでき、計算資源を最大活用できます。 - (2)がん全ゲノム解析の高精度・標準化パイプラインの構築
がん全ゲノムでの変異の結果は、ゲノム解析アルゴリズムによって大きく異なります。PCAWGでは、三つのゲノムセンターの基本解析パイプラインを組みわせて、高精度で標準化された変異同定解析手法を構築しました。この方法を用いて、(1)の仮想巨大データセンター上で2,834例のがん全ゲノムシーケンスデータ解析を行い、網羅的かつ精度の高い変異データセットを作成しました。そして、このデータをPCAWG参加研究者と仮想クラウド上において共有しました。 - (3)変異・異常の特徴を解明するワーキンググループの結成
約30億塩基対からなるヒトゲノムを偏りなくシーケンス解析することで、非コード領域の変異、ゲノム構造異常、ミトコンドリアゲノム異常、がん内に潜むウイルスなど病原体の検出、テロメア[12]の長さの異常などの特徴を見つけることが可能です。そのために、特徴ごとに16のワーキンググループを結成し、国際共同で解析を行いました(図2)。
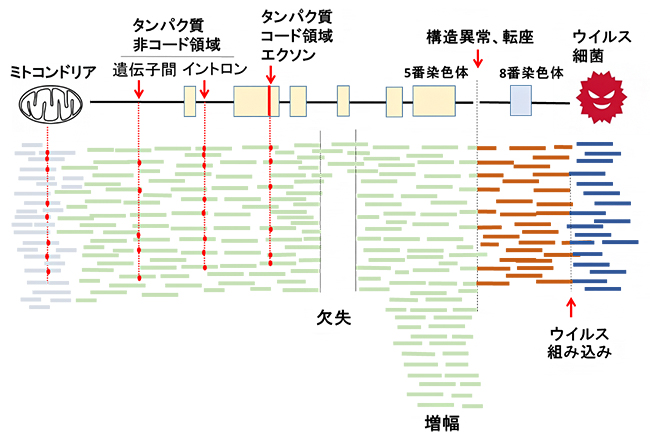
図2 全ゲノムシーケンス解析によって見いだされる変異
30億塩基対からなるヒトゲノムを偏りなくシーケンス解析することによって、ミトコンドリアゲノム異常、非コード領域の変異、コピー数異常(欠失、増幅)、ゲノム構造異常、ウイルスの検出などが可能。図の横棒は100個ほどの塩基配列(シーケンス)を示し、赤の点は変異を表している。
今回のがん全ゲノムの大規模解析では、がんパネル検査やエクソーム解析からの一部検出とは異なり、ほとんど全てのがんでの変異・異常が検出できます。解析の結果、4,400万個の一塩基変異、240万個の短い欠失・挿入、29万個の構造異常、約8,000個のミトコンドリアゲノム異常、テロメア/TERT遺伝子異常など、合計で4,600万個を超える変異・異常が同定され(図3左)、さらにそれらのさまざまな特徴が明らかになりました。例えば、がん体細胞の一塩基変異数は、がんの診断年齢が高いほど多い傾向にありました(図3右)。こうして、これまでで最も網羅的かつ詳細ながんゲノムマップを作成することができました(図4)。
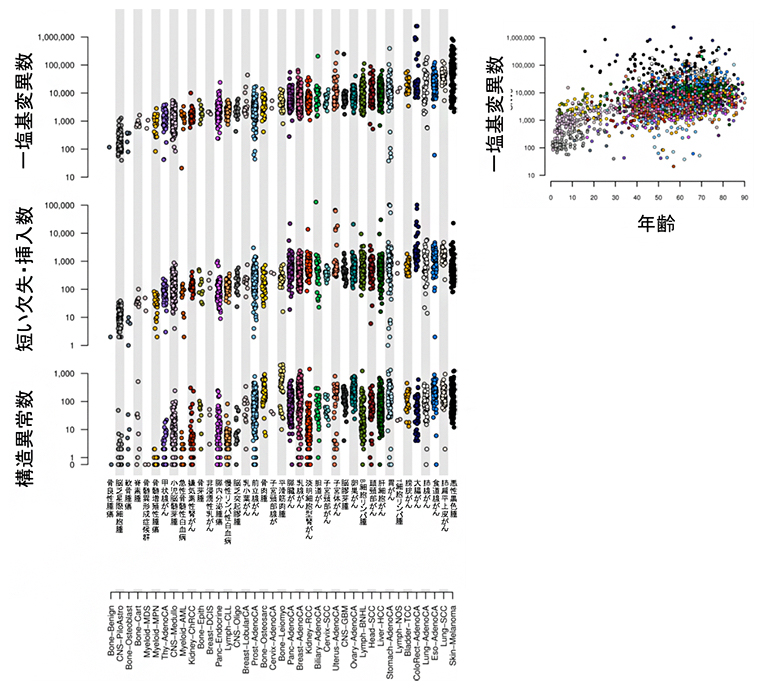
図3 全ゲノム上での各種がんの変異・異常
- 左:38種類のがんについて、約4,400万個の一塩基変異、約240万個の短い欠失・挿入、約29万個の構造異常、約8,000個のミトコンドリアゲノム変異を同定した。1つの点が1つの腫瘍の変異数を示している。
- 右:がん体細胞の変異数は、がんの診断年齢が高いほど多い傾向にあった。
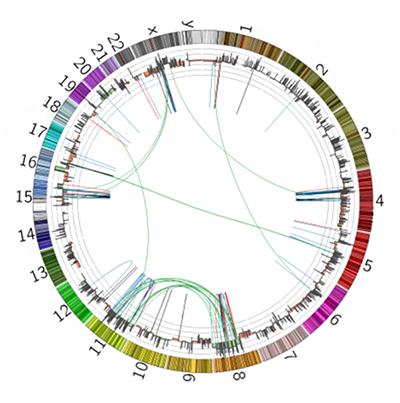
図4 全ゲノムでの変異マップの肝臓がんの一例
1番から22番染色体、X染色体Y染色体を環状に並べて、肝臓がんに関して全ゲノムレベルでの異常を示している。8番染色体と11番染色体の間で、多数の転座(ゲノム構造異常)が見られる。
今後の期待
今後、DNAシーケンス解析技術の革新に伴うコストの低下により、全ゲノムシーケンス解析が、研究分野のみならず、がんの診断や個別化医療の分野においても、標準的なゲノム解析手法になると予測されています。
PCAWGでの全ゲノムデータや開発した解析手法は、世界中で公開されています。PCAWGでのがんシーケンス解析の経験と手法、データセットは、次世代のがんゲノム医療および研究のデータ基盤になると期待できます。
補足説明
- 1.全ゲノムシーケンス解析
次世代シーケンサーを使って、個人(約30億塩基対)やがんの全ゲノム情報を解読し、塩基配列の違いや変化を同定すること。データが大量になるため、大型計算機を使って情報解析を行うのが一般的である。タンパク質をコードする1~2%の範囲のエクソンだけでなく、遺伝子の発現を制御するゲノム領域の変異やさまざまな構造異常(大きなゲノム配列異常)も検出可能で、究極のゲノム解析手法といえる。がんの場合は、がんのDNAと同一患者由来の正常DNAの全ゲノムシーケンス解析を行い、その差分を調べる。超大量の情報を扱うため、多大な労力と時間を要し、高度な数学や統計学、遺伝学、情報工学の知識と技術が必要である。この情報解析の方法によって、変異の結果が大きく異なる場合があるため、解析の標準化が求められている。 - 2.全ゲノム横断的がん解析プロジェクト(PCAWG)
ICGC/TCGA内のがんの全ゲノムシーケンス解析のデータを集積し、ICGC/TCGAの共同作業にてがんの横断的(PanCancer)解析を行うプロジェクトとして、2014年に始動した。約2,800例のさまざまながんの全ゲノムシーケンスのビックデータを東京大学医科学研究所ヒトゲノム解析センターのスーパーコンピュータ「SHIROKANE」を含む世界10カ所のデータセンターで仮想空間を作り、分担して解析を行っている。生シークエンスデータだけで約1ペタバイト(1000兆バイト)の情報量になる。PCAWGは、PanCancer Analysis of Whole Genomesの略。 - 3.非コード領域
ヒトゲノム(約30億塩基対)のうち、タンパク質をコードしている遺伝子領域はわずか1~2%であり、残りの領域は非コード領域と呼ばれる。非コード領域には、遺伝子発現を制御している領域や、タンパク質に翻訳されないRNA(非コードRNA)をコードしている領域が含まれ、遺伝子の転写調節やゲノムの複雑な構造を調節していると考えられている。 - 4.ゲノム構造異常
1塩基の配列が変化する点突然変異と異なり、数百~数百万塩基の配列が欠失、組み込み、重複、逆位(方向が逆になる)、染色体間で転座する(移動する)など、大きなゲノム配列の変化をいう。全ゲノムシーケンス解析にて、網羅的に検出できるようになった。 - 5.ミトコンドリアゲノム
ミトコンドリアは、細胞に必要なエネルギーを取り出す呼吸機能を担う細胞内小器官。ミトコンドリアは細胞内に数百個以上存在し、ミトコンドリア内にも16,000塩基対ほどのゲノムが存在する。ミトコンドリアゲノムは、呼吸機能をつかさどる酵素など37個の遺伝子をコードしている。 - 6.がんゲノムマップ
30億塩基からなるヒトゲノムマップで、どの位置にどういった型の変異が蓄積しているのかを示す。例えば、最も多く見つかるKRAS遺伝子の変異は、12番染色体の25245350の位置にCからTへの変異が見られる。 - 7.国際がんゲノムコンソーシアム(ICGC)
がんのゲノム異常の包括的なカタログを作成する目的のため、2008年に発足した国際連携研究組織。ICGCの各メンバーは、データ収集・解析に関するICGCの共通基準の下、1種類のがんについて500症例を解析し、ICGCのデータベースに登録して世界中に公開する。2015年時点で、米国の大規模がんゲノムプロジェクト(TCGA)に加えて、ヨーロッパ、南北アメリカ、アジア、オーストラリアの16カ国およびEUの機関や組織が参画し、25,000例のエクソームを主としたゲノム変異データが蓄積された。このデータを公開した2018年に終了。ICGCはInternational Cancer Genome Consortiumの略。 - 8.TCGA
がんゲノムアトラス計画という米国主導の大規模ながんゲノム解析プロジェクト。ゲノム、トランスクリプトーム、エピジェネティックな修飾など、がんゲノムについて包括的な理解を目指し解析を進めている。TCGAはThe Cancer Genome Atlasの略。 - 9.次世代シーケンサー
ヒトゲノムの全配列を1,000米ドル以下のコストで解読すべく、欧米の政府や企業が技術開発を行った結果、より高速高精度の性能を持つシーケンサーが開発された。従来の方法に比べ、超大量のDNAシーケンス反応を並列して行うことができる。 - 10.パイプライン
ゲノムなどの大量のデータを解析するための中心的なアルゴリズム。がんの全ゲノムシークエンスデータから変異を同定するパイプラインは、各々のゲノムセンターが独自のパイプラインを持っており、同一のデータからでも結果が異なることが分かっている。 - 11.仮想マシーン(VM)
複数の物理サーバーを一つの大きなサーバーに見立て、統一した計算スペースを確保する、または一つの物理サーバーを複数に分割して異なるOSで走らせることができるコンピューター技術。限られた容量の物理サーバーを効率良く使うことができる。VM はVirtual Machineの略。 - 12.テロメア
細胞分裂で染色体(DNA)が複製されるとき、その機能上どうしても複製できない部分が末端に生じてしまう。それを補うためにテロメアと呼ばれる長い繰り返し配列が染色体の末端に作られており、細胞分裂をするたびに少しずつ短くなっていく。テロメアの長さを調べると細胞分裂をした回数が分かることから、ある程度細胞の年齢を推測できる。TERT(Telomerase Reverse Transcriptase)遺伝子は、このテロメアの長さを維持するために必要なテロメア逆転写酵素をコードしている。
国際共同研究グループ
本研究は、国際的連携による全ゲノムがん種横断的解析プロジェクト(PCAWG)の一環として行い、国際共同研究グループ「The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium」には、37カ国から1,300人以上が参加しました。日本からの参加者は下記の通り。
理化学研究所 生命医科学研究センター
がんゲノム研究チーム
チームリーダー 中川 英刀(なかがわ ひでわき)
副チームリーダー(研究当時) 藤本 明洋(ふじもと あきひろ)
上級研究員藤田征志(ふじた まさし)
医科学数理研究チーム
チームリーダー 角田 達彦(つのだ たつひこ)
東京大学 医科学研究所
ヒトゲノム解析センター DNA情報解析分野
助教(研究当時) 白石 友一(しらいし ゆういち)
教授 宮野 悟(みやの さとる)
ヘルスインテリジェンスセンター 健康医療データサイエンス分野
教授 井元 清哉(いもと せいや)
原論文情報
本研究を含むPCAWGの研究成果は、Nature誌の特集号として『Nature』に6報、『Nature Genetics』に5報、『Nature Communications』に9報、『Nature Biotech』に1報などが掲載されます。本研究に特に関係する論文は下記の通りです。
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium, "Pan-cancer analysis of whole genomes", Nature, 10.1038/s41586-020-1969-6
- Esther Rheinbay, et al., "Analyses of non-coding somatic drivers in 2,658 cancer whole genomes", Nature, 10.1038/s41586-020-1965-x
- Nicola D Roberts, et al., "Patterns of somatic structural variation in human cancer genomes", Nature, 10.1038/s41586-019-1913-9
- Yuan Yuan, et al., "Comprehensive Molecular Characterization of Mitochondrial Genomes in Human Cancers", Nature Genetics, 10.1038/s41588-019-0557-x
発表者
理化学研究所
統合生命医科学研究センター がんゲノム研究チーム
チームリーダー 中川 英刀(なかがわ ひでわき)
東京大学 医科学研究所 ヘルスインテリジェンスセンター
健康医療データサイエンス分野
教授 井元 清哉(いもと せいや)
報道担当
理化学研究所 広報室 報道担当
お問い合わせフォーム
東京大学医科学研究所 管理課 総務チーム
Tel: 03-6409-2018 / Fax: 03-5449-5402
E-mail: koho [at] ims.u-tokyo.ac.jp ※[at]は@に置き換えてください。