2019年2月25日
理化学研究所
東京大学大学院農学生命科学研究科
日本医療研究開発機構
高速検索エンジン「CellFishing.jl」を開発
-大規模1細胞データベースから類似細胞を瞬時に検出する手法-
理化学研究所(理研)生命機能科学研究センターバイオインフォマティクス研究開発ユニットの佐藤建太研究パートタイマーI(東京大学大学院農学生命科学研究科)、露崎弘毅特別研究員、二階堂愛ユニットリーダー、東京大学大学院農学生命科学研究科の清水謙多郎教授の共同研究チームは、大規模1細胞データベース(DB)から、類似細胞を高速検索するソフトウェア「CellFishing.jl」を開発しました。
本研究成果は、細胞分化や臓器・器官発生などの基礎研究から、再生医療における移植細胞の有効性・安全性評価、創薬などの発展に貢献すると期待できます。
多細胞生物が持つ数百種類の細胞の機能を理解する方法として、1細胞ごとにRNAの種類と量を計測する「1細胞RNAシーケンス法[1]」があります。これにより、共同研究チームを含め、世界中が協力してヒトの全細胞種の遺伝子発現データをデータベース化する研究が進行中で、次々とデータが公開されています。世界中の研究者がこれらのDBと自分のデータを比較できれば、疾患の原因や薬剤応答を細胞レベルで精緻に理解できるようになります。
今回、共同研究チームは、1細胞発現データを検索するアルゴリズムを開発し、百万個の細胞DBから、高い検索精度を保ちつつも、1細胞あたり0.63ミリ秒で類似細胞の検索に成功しました。これは既存の方法の100倍以上の速さです。さらに、類似細胞で特徴的に働く遺伝子を高速に検出する機能も開発しました。
本研究は、英国の科学雑誌『Genome Biology』(2月11日付)に掲載されました。
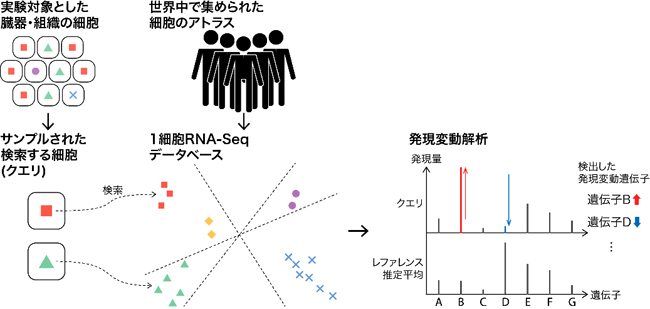
図 細胞検索と発現変動解析
※研究支援
本研究の一部は、科学技術振興機構(JST)戦略的創造研究推進事業(CREST)「[1細胞] 統合1細胞解析のための革新的技術基盤(研究統括:菅野純夫)」の研究課題「臓器・組織内未知細胞の命運・機能の1細胞オミクス同時計測(研究代表者: 二階堂愛)」、JSTおよび日本医療研究開発機構(AMED)再生医療実現拠点ネットワークプログラム「iPS・分化細胞集団の不均質性を1細胞・全遺伝子解像度で高速に測定する技術の開発(研究代表者: 二階堂愛)」「超多検体オミクスによる細胞特性の計測(研究代表者: 二階堂愛)」、日本学術振興会(JSPS)科学研究費補助金若手研究(B)「1細胞RNA-Seqデータ内に含まれる細胞型を特定する解析手法の確立(研究代表者:露崎 弘毅)」の支援を受けて行われました。
背景
私たちの臓器には多種多様な細胞が含まれており、これらの細胞が互いに関連して臓器の機能を支えています。しかし、臓器にどのような細胞がどの程度含まれているか、それぞれの細胞がどのように臓器機能を支えているかについては、十分に理解が進んでいません。臓器の疾患の理解や診断、創薬を精緻に行うためには、臓器に含まれる細胞種類[2]やその数、細胞状態[2]やその機能、薬剤に対する応答を調べる必要があります。
細胞が持つ多様な機能は、ゲノムDNAにコードされた数万種類のRNAの組み合わせによって決まります。RNAは、さまざまなタンパク質に翻訳され、細胞のさまざまな機能を担います。従って、臓器を構成する細胞種を判別し、その機能を類推するには、1細胞が持つRNAの量と種類(1細胞遺伝子発現プロファイル[3])を知る必要があります。これを実現する技術が、「1細胞RNAシーケンス法(1細胞RNA-seq)」です。
臓器内の多種多様な細胞集団や希少な細胞を漏れなく捉えるには、一度の実験で大量の1細胞RNA-seqを実施する必要があります。これを実現するのが、「高出力型1細胞RNA-seq法[4]」です。二階堂ユニットリーダーらは、これまで高出力型1細胞RNA-seq法Quartz-Seq2注1,2)や、1細胞完全長トータルRNAシーケンス法RamDA-seq注3)など実験技術の開発で世界をリードしてきました。
高出力型1細胞RNA-seqの登場により、世界中の研究者がさまざまな細胞の1細胞遺伝子発現プロファイルを計測できるようになり、これらのデータは日々インターネット上に公開されています。その主な例として、現在進行中の国際共同プロジェクト「ヒト細胞アトラス(HCA)[5]」が挙げられます。HCAでは、ヒト細胞全種類の1細胞遺伝子発現プロファイル「全細胞アトラス」[6]の構築が試みられており、全細胞アトラスの完成によって疾患の解明や創薬技術などが進展すると期待されています。日本からは、本共同研究チームや理研生命医科学研究センター(IMS)など注4)が参加しています。
このような状況で、世界中の研究者が自分の研究で得た1細胞遺伝子発現プロファイルと大規模データとを比較することで、例えば、疾患に関わる細胞や遺伝子を発見できると期待されています。一般に、1細胞遺伝子発現プロファイルが似ていれば、その細胞の機能や状態も似ていると考えられます。また、似た細胞を検索できれば、正常細胞と疾患細胞(1細胞遺伝子発現プロファイルがわずかに異なる細胞)を発見し、その違いを研究することもできます。
しかし、計測される1細胞遺伝子発現プロファイルも、データベースもそれぞれ巨大であるため、高速に、かつ正確に、1細胞遺伝子発現プロファイルが似た細胞を検索するアルゴリズムが必要とされていました。このような背景から、研究者が得た1細胞RNA-seqデータを既存のデータセットに対して、高速に検索する手法の開発が求められていました。
- 注1) 2013年7月25日プレスリリース「細胞1個の遺伝子発現を網羅的に定量化する「Quartz-Seq法」を開発」
- 注2) 2018年3月13日プレスリリース「数千個の1細胞からRNA量と種類を正確に計測」
- 注3) 2018年2月14日プレスリリース「1細胞から多種多様なRNAのふるまいを計測」
- 注4) 2017年12月20日ブログ「アジア初のHuman Cell Atlas会議が開催されました」
研究手法と成果
共同研究チームは、大規模な全細胞アトラスから検索用データベースを構築し、そのデータベースから遺伝子発現パターンが類似した細胞を取得するソフトウェア「CellFishing.jl(セルフィッシングジェーエル)」を開発しました。
CellFishing.jlは、全細胞アトラスが持つ巨大な1細胞遺伝子発現データからのデータベース構築と、そのデータベースでの検索という2つの機能を持ちます。データベース構築については、アトラスにある全細胞(レファレンス細胞)の1細胞遺伝子発現プロファイルをLocality Sensitive Hashing[7]という手法で0と1からなるビット列の表現に圧縮します。この圧縮データに、高速検索用の索引をつけて、データベースを構築します。共同研究チームが開発した新しい索引技術により、高速なデータの取り出しが可能になります。
データベース検索については、まず、ユーザーが検索する細胞(クエリ細胞)を、データベースと同じようにビット列に変換します。次に、ビット列間の類似度を基に、1細胞遺伝子発現プロファイルが類似したレファレンス細胞をデータベースから探します(図1)。これらの計算は、一般的なCPUに実装されている命令を最大限に活用して実装することで、GPUなどの特殊な装置を持たない一般的な計算機でも、高速に検索できるようになりました。
共同研究チームは、CellFishing.jlの検索精度を評価するため、専門家による細胞型の注釈が付けられている四つの1細胞RNA-seqデータセットを用いて、クエリ細胞と、検索した結果得られた細胞の細胞型の一致率を交差検定[8]で評価したところ、実験したほぼ全てのケースにおいて、既存の手法と比較して、0.5%から5%ほどの一致率の向上が見られました(図2左)。
また、実行にかかる時間を測定したところ、同じ既存手法と比較して最大のデータセットではデータベースの構築時間が約22分の1に、検索時間が約118分の1に短縮されました(図2右)。また、存在比率が0.1%を下回るような希少な細胞型についても比較的高い一致率が得られていることが分かりました。さらに、独立した実験から得られたデータセットを検索した場合でも、高い一致率を維持することが分かりました。
次に、1細胞遺伝子発現プロファイルが似て非なる細胞を発見し、その原因となる遺伝子を特定する機能を実装しました。この機能によって、同じ細胞種でありながら、機能が未成熟である細胞と成熟した細胞、あるいは、正常細胞と疾患を発症した細胞などを検索し、その原因遺伝子を発見できます。
まず、類似した細胞を発見した後、類似細胞で特徴的に働いている遺伝子を瞬時に検出する手法を実装しました。これを評価するために、マウスの骨髄から得られた未成熟B細胞をクエリ細胞として、マウス全細胞アトラスに対して検索したところ、発達段階の異なる3種類のB細胞を発見できました。さらに、それらについて発現変動遺伝子を検出したところ、B細胞の成熟と関連があると考えられる遺伝子が複数得られました。これにより、似て非なる細胞を高速に検索した後、その原因となる遺伝子の候補も瞬時に得られることを示しました。このような機能は既存の検索エンジンには実装されておらず、CellFishing.jlの画期的な機能の1つです。
続いて、開発した手法が大規模なデータベースで利用できることを実証する実験を行いました。現時点で入手できる最も大きな1細胞RNA-seqのデータセットを用い、レファレンス細胞の数を213(≒8,200)から220(≒105万)まで2倍ずつ8段階で大きくしながら検索用データベースを構築し、別に用意した10,000細胞を検索しました。データベースの構築時間は細胞数とよく比例し、最大サイズでも125.0秒ほどでした。細胞の検索時間は最大サイズで6.3秒(1細胞あたり0.63ミリ秒)、データベースのメモリサイズは183.3メビバイト(MiB)[9]でした。検索時間の増加はレファレンス細胞の数の増加よりも緩やかで、メモリサイズも小さいことから、今後さらに大規模なデータセットへの適用も期待できます。
CellFishing.jlはすでにオープンソース・ソフトウェアとして公開されており、実装に使ったJulia言語[10]と併せて、誰でも自由に使用できます。
今後の期待
本研究では、遺伝子発現の類似した細胞を検索でき、かつ大規模なデータセットにも適用可能な手法の開発・実証に成功しました。この成果は、細胞分化や臓器・器官発生などの基礎研究から、再生医療における移植細胞の有効性・安全性評価など、さまざまなライフサイエンスの研究分野の発展への貢献が期待できます。例えば、移植用にiPS細胞[11]から目的の分化細胞を作製する再生医療では、分化細胞が正しく分化しているかを判断するために、正常なヒト細胞アトラスを検索し、分化細胞との類似度を調べる必要があります。
また、創薬分野では、疾患のある細胞を正常細胞に戻す化合物発見に貢献すると期待できます。例えば、疾患のある細胞に薬剤を加えて1細胞RNA-seqを実施し、そのデータをヒト健常細胞アトラスと比較することで、細胞がどの程度正常になったか評価できます。
今後、1細胞アトラスの整備が進めば、数億の1細胞データがアトラス化されると見込まれます。巨大化する1細胞アトラスを高速かつ正確に検索するために、アルゴリズムのさらなる開発が望まれます。
原論文情報
- Kenta Sato, Koki Tsuyuzaki, Kentaro Shimizu, Itoshi Nikaido, "CellFishing.jl: an ultrafast and scalable cell search method for single-cell RNA-sequencing", Genome Biology, 10.1186/s13059-019-1639-x
発表者
理化学研究所
生命機能科学研究センター バイオインフォマティクス研究開発ユニット
研究パートタイマーⅠ 佐藤 建太(さとう けんた)
特別研究員 露崎 弘毅(つゆざき こうき)
ユニットリーダー 二階堂 愛(にかいどう いとし)
東京大学大学院農学生命科学研究科
応用生命工学専攻 生物情報工学研究室
教授 清水 謙多郎(しみず けんたろう)
報道担当
理化学研究所 広報室 報道担当
Tel: 048-467-9272 / Fax: 048-462-4715
お問い合わせフォーム
東京大学農学系事務部 総務課総務チーム 広報情報担当
Tel: 03-5841-5484 / Fax: 03-5841-5028
E-mail:koho [at] ofc.a.u-tokyo.ac.jp
※上記の[at]は@に置き換えてください。
産業利用に関するお問い合わせ
AMED事業に関するお問い合わせ先
国立研究開発法人日本医療研究開発機構(AMED)
戦略推進部 再生医療研究課
〒100-0004 東京都千代田区大手町一丁目7番1号
Tel: 03-6870-2220 / Fax: 03-6870-2242
E-mail:saisei[at]amed.go.jp
※上記の[at]は@に置き換えてください。
補足説明
- 1.1細胞RNAシーケンス法(1細胞RNA-seq)
1細胞中に含まれるRNAをDNAシーケンサーで配列決定し、網羅的かつ定量的にその量や種類を決定する方法。微量なRNAを用いるため、微量RNAから相補的DNA(cDNA)を合成する「逆転写反応」と、シーケンス可能な量までcDNAを増幅させる「全cDNA増幅法」の二つのステップからなる。 - 2.細胞種類、細胞状態
多細胞生物は、たくさんの細胞から構成される。それぞれの細胞は役割、すなわち機能を持つ。例えば、免疫を担う細胞や角膜のように透明な細胞、神経活動を支える電気や化学物質でほかの細胞と通信する細胞などがある。また、同じ細胞でもその状態が異なる場合がある。免疫の細胞でも機能が未熟なものから、成熟して機能を発揮する細胞まである。 - 3.1細胞発現プロファイル
細胞が持つRNAの種類とその量を数値で表したもの。通常の1細胞では、数万遺伝子のうち数百~数千の遺伝子からRNAが転写されており、遺伝子によるが、数分子から数千分子程度のRNAが細胞内に存在する。このプロファイルが似ていれば、細胞の機能も似ていると考えることができる。1細胞トランスクリプトーム、1細胞遺伝子発現パターンと呼ぶこともある。 - 4.高出力型1細胞RNA-seq法
数千から数万個の1細胞を採取し、1細胞RNA-seqを実施する技術。1細胞から取り出したRNAに細胞を認識するDNA配列(DNAバーコード)を付加し、混合反応することで、少ない試薬・手順で、シーケンスが可能になる。シーケンスデータに含まれるDNAバーコードをもとにデータを1細胞ごとのデータに分離する。 - 5.ヒト細胞アトラス(HCA)
2016年にスタートした、ヒトが持つ全細胞種類の1遺伝子発現地図(アトラス)を計測しデータベース化する国際研究プロジェクト。HCAはHuman Cell Atlasの略。 - 6.全細胞アトラス
1細胞RNA-seqを用いて、ある生物の全細胞種類の遺伝子発現をデータベース化したもの、あるいは、そのプロジェクト。数百万から数億細胞×数万遺伝子のRNA量の行列データになると見込まれる。ある生物の全細胞のデータベースを、その生物の全細胞の「地図(アトラス)」になぞらえた呼び方。 - 7.Locality Sensitive Hashing
データ点が存在する空間をランダムに分割し、データ点の座標を0と1からなるビット列に圧縮してデータ点間の類似度を近似する手法。 - 8.交差検定
機械学習の性能を評価する方法の一つ。用意したデータをいくつかのデータに分け、そのうちの一つをテストデータ、それ以外を学習データとして学習し、学習法を評価する方法。クロスバリデーションともいう。 - 9.メビバイト(MiB)
コンピュータの記憶容量の大きさの単位。1MiBは220=1,048,576バイトを意味する。 - 10.Julia言語
2012年に公開された比較的新しいプログラミング言語。高速に動作し、科学技術計算に使われるさまざまな機能を標準で備えるため、ライフサイエンスのみならず、データサイエンス分野で広く利用されつつある。 - 11.iPS細胞
人の皮膚細胞などの体細胞・組織から採取した細胞にOct4、Sox2、c-Myc、Klf4遺伝子などを導入して初期化し多能性を持たせ、人工的に作製した多能性幹細胞。iPSはInduced Pluripotent Stemの略。
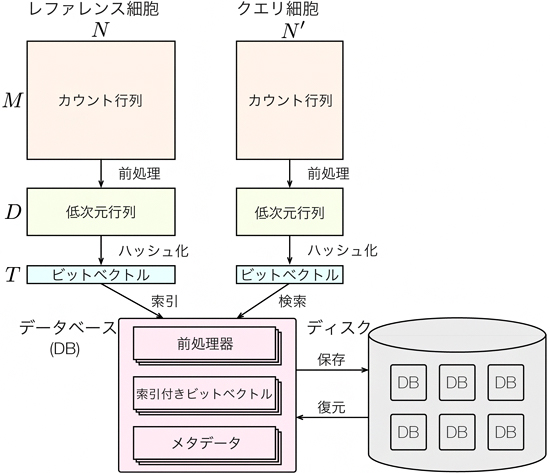
図1 CellFishing.jlの模式図
CellFishing.jlには、アトラスにある全細胞からデータベース(DB)を構築し、ユーザーが検索するクエリ細胞を検索する2つの機能がある。データベース自体はファイルに保存することもできる。
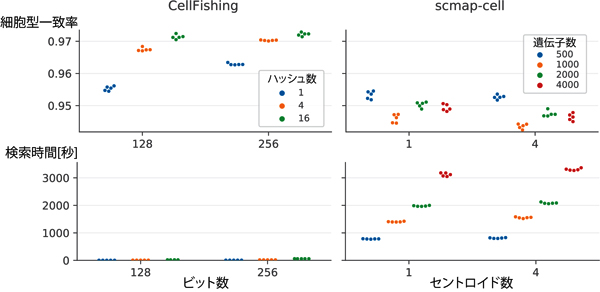
図2 検索精度(上)と検索時間(下)
CellFishing.jlと既存手法(scmap)を、パラメータを変えながら検索精度と検索時間の両面で比較。上段のパネルの縦軸は、検索精度(正解との細胞型一致率)を示し、数値が高いほど正確に検索できたことを示す。下段の縦軸は検索時間(秒)を示し、数値が低いほど高速であることを示す。各パネルの横軸は、検索する際の計算条件を示す。どの計算条件でも、検索精度と速度ともにCellFishing.jlが勝っているのが分かる。