2022年4月12日
理化学研究所
日本医療研究開発機構
次世代天然物化学技術研究組合
アロステリック薬剤はタンパク質の構造平衡を変化させる
-既存のGPCR標的薬の限界を克服する新薬開発に期待-
理化学研究所(理研)生命機能科学研究センター生体分子動的構造研究チームの嶋田一夫チームリーダー(次世代天然物化学技術研究組合技術顧問)、今井駿輔上級研究員、金子舜研修生らの共同研究チームは、Gタンパク質共役型受容体(GPCR)[1]を標的とする既存の医薬品の薬効が、新たなGPCR標的薬として期待されているアロステリックモジュレーター(アロステリック薬剤)[2]により高められる仕組みを明らかにしました。
本研究成果は、GPCRを標的としたアロステリック薬剤の合理的な設計や、既存品より高い薬効度を持つ医薬品の開発に貢献すると期待できます。
膜タンパク質であるGPCRを標的とする医薬品は、GPCRの細胞外側に存在するポケットに結合してGPCRを活性化します。しかし、既存薬はGPCRの活性を十分に引き出すことができないことから、このポケット以外の場所に結合して作用するアロステリック薬剤の実用化が期待されています。
今回、共同研究チームは、溶液核磁気共鳴(NMR)法[3]を用いた立体構造解析により、GPCRの一種μオピオイド受容体(MOR)[4]に対するアロステリック薬剤の活性化機構を調べました。その結果、MORは活性のない不活性化型、活性の低い部分活性化型、活性の最も高い完全活性化型の三つの構造を行き来する平衡状態(構造平衡)にあり、各構造の存在比がMORの活性を決定していることが分かりました。既存薬が単独で結合しても完全活性化型構造の存在比は低い一方、アロステリック薬剤が同時に結合すると完全活性化型構造の存在比が増加し、MORの活性もさらに上昇することが明らかになりました。
本研究は、科学雑誌『Proceedings of the National Academy of Sciences of the United States of America(PNAS)』オンライン版(4月11日付:日本時間4月12日)に掲載されました。
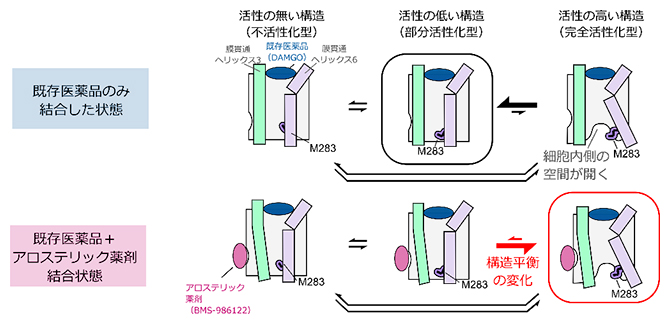
アロステリック薬剤によるGタンパク質共役型受容体の構造平衡の変化
背景
Gタンパク質共役型受容体(GPCR)は、米国食品医薬品局(FDA)承認薬の標的の30%以上を占める、創薬標的として極めて重要な膜タンパク質ファミリーです。GPCRは細胞外のさまざまな刺激を細胞内のGタンパク質[1]に伝える機能を持ち、その最初のステップの一つは、ホルモンなどの分泌因子がGPCRの細胞外側に存在するポケットに結合することです。GPCRを標的とした医薬品の多くは、分泌因子の代わりにこのポケットに結合し、GPCRを活性化することによりシグナルを伝達します。この際に伝達されるシグナルの強さにより医薬品の薬効度が決定すると考えられており、GPCRのシグナル伝達活性を増加させることで、より強い薬効を持つ医薬品が開発できると期待されます。
しかし、細胞外側のポケットを標的とした化合物では、既存薬より強くGPCRを活性化するものが新たに見つかることはまれであり、GPCRの細胞外側ポケットを標的とした医薬品の薬効度の上限をいかに克服するかが課題となっています。
一方で、細胞外側ポケットとは異なる部位に結合する「アロステリックモジュレーター(アロステリック薬剤)」と呼ばれる新しいクラスの薬剤が報告されています。これは、細胞外側ポケットに結合する医薬品と、ポケットとは異なる場所に結合するアロステリック薬剤が同時に存在すると、GPCRのシグナル伝達活性が増強されるという現象を応用したものです。アロステリック薬剤は、既存薬の薬効度の限界を打ち破る新たな医薬品候補として期待されています。
GPCRに対するアロステリック薬剤の詳しい作用機構を調べるため、両者が結合した状態の構造解析が複数報告されています。しかしこれまでの解析は、時間軸を考慮しない静的な条件で行われており、アロステリック薬剤の結合に伴うGPCRの構造変化は、ほとんど観測されていませんでした。そのため、なぜアロステリック薬剤がGPCRのシグナル伝達活性を増強できるのか、その機構は理解されておらず、合理的なアロステリック薬剤の設計には至っていませんでした。
研究手法と成果
今回、共同研究チームは、鎮痛作用に関わる代表的なGPCRの一種である「μ(ミュー)オピオイド受容体(MOR)」を対象に、アロステリック薬剤である化合物BMS-986122(図1A)による活性増強機構をMORの動的な構造変化に着目して解析しました。
まず、アロステリック薬剤によってMORのシグナル伝達活性がどの程度上昇するかを解析しました。その結果、BMS-986122の結合に伴い、MORのシグナル伝達活性が増加することが確かめられました。特に、これまでに報告されている作動薬の中で最も強い薬効度を持つことが知られている薬剤DAMGOが結合した状態でも、BMS-986122の結合によってMORのシグナル伝達活性がさらに1.7倍にまで上昇しました(図1B)。このことから、BMS-986122を併用することで、既存薬が単独で結合した際よりも強くMORを活性化できることが示されました。
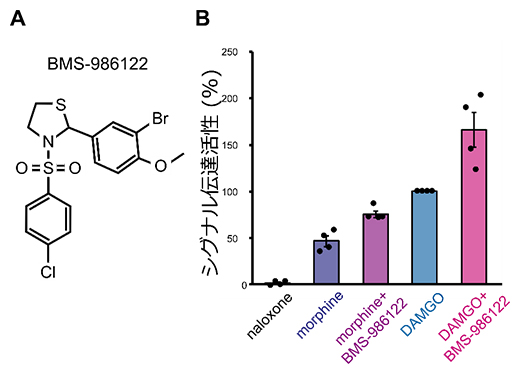
図1 アロステリック薬剤がMORのシグナル伝達活性に与える影響
- A.アロステリック薬剤BMS-986122の化学構造式。
- B.MORのシグナル伝達活性を解析したグラフ。シグナル伝達活性は、MORにより細胞内で活性化されるGタンパク質のGTP加水分解量の測定により解析した。MORの活性を阻害する拮抗薬(naloxone)に対して、鎮痛作用を示す部分作動薬(morphine)、既存薬で薬効度の最も高い完全作動薬(DAMGO)それぞれの結合状態における、アロステリック薬剤(BMS-986122)非存在下または存在下でのMORのシグナル伝達活性を比較した。縦軸は、DAMGO結合状態MORのシグナル伝達活性を100%として規格化した数値。BMS-986122が作動薬によるシグナル伝達活性をさらに上昇させることが示された。
次に、BMS-986122結合に伴い、MORにどのような構造変化が生じるかを溶液核磁気共鳴(NMR)法により調べました。MORのアミノ酸配列のうち283番目にあるメチオニン(M283)は、Gタンパク質との結合領域の中でも細胞質側に突き出た位置にあるため、MORの構造変化を反映したNMRシグナルが得られると期待されます。そこで、さまざまな医薬品が結合したMORについて、BMS-986122結合前後でのNMRスペクトルを比較したところ、M283のNMRシグナルが最大三つに分裂して観測され、各NMRシグナルの存在比が条件によって変化していることが分かりました(図2)。
この存在比と上記で測定したGタンパク質シグナル伝達活性を比較したところ、この三つのNMRシグナルはそれぞれ、シグナル伝達活性のない「不活性化型」、シグナル伝達活性の低い「部分活性化型」、シグナル伝達活性の高い「完全活性化型」の構造を反映していることが示されました(図2)。特に、既存の作動薬DAMGOが単独で結合した状態では完全活性化型構造の存在比が19%と低かった一方で、BMS-986122がさらに結合すると完全活性化型構造の存在比が73%に増加していることが明らかになりました。このことは、BMS-986122がMORの構造平衡中の各構造の存在比を変えることで、シグナル伝達活性の増強を引き起こしていることを示しています。
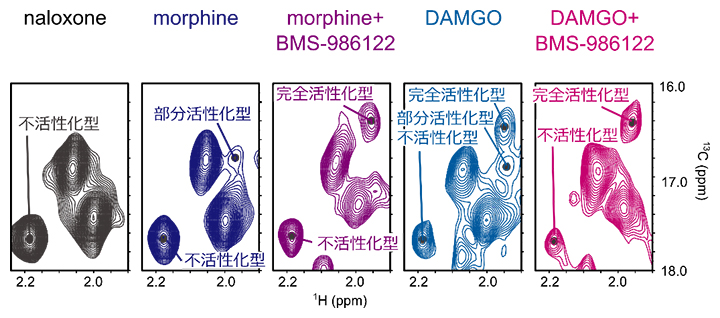
図2 MORの三つの活性化型構造を反映するM283のNMRシグナル
拮抗薬(naloxone)に対して、部分作動薬(morphine)、完全作動薬(DAMGO)結合状態におけるアロステリック薬剤(BMS-986122)非存在下または存在下でのMORのM283のNMRシグナルを2次元のスペクトルで比較したグラフ。BMS-986122が存在すると、MORの完全活性型構造を示すNMRシグナルが増加することが示された。
次に、不活性化型・部分活性化型・完全活性化型のMORの構造がどのように異なるのかを調べるために、溶液NMR法によるさらなる解析を行いました。タンパク質に構造変化が生じると、タンパク質を構成するアミノ酸と、タンパク質の周囲にある溶液や溶媒中の分子との相互作用も変化します。この変化は、常磁性錯体[5]であるGd-DTPA-BMAを溶媒に添加すると、常磁性緩和促進効果[5]によるNMRシグナル強度の変化として捉えることができます。
この条件でのNMR解析を行った結果、M283の三つのNMRシグナルのうち、完全活性化型を反映するNMRシグナルでのみ、NMRシグナル強度の著しい減弱が観測されました(図3A)。これは、完全活性化型構造でのみM283がGd-DTPA-BMAと近接したことを意味し、M283のNMRシグナルに反映される三つの構造のうち、完全活性化型構造でのみM283が溶媒へ露出していることが示されました。
MORをはじめとしたGPCRは一般的に、Gタンパク質の結合に伴って細胞内側に空間が開くことが知られており、M283はその空間が開くことにより溶媒に露出する領域に位置しています。このことから、完全活性化型でのM283の溶媒露出度の増加は、完全活性化型構造において細胞内側に空間が開いていることを示します(図3B)。
以上の結果から、MORは細胞内側が閉じた不活性化型、部分活性化型、および細胞内側が開いた完全活性化型の三つの構造を行き来する構造平衡にあり、その存在比がMORのシグナル伝達活性を規定していることが明らかになりました(図3C)。既存薬の中で最も強い薬効度を示す化合物が結合した状態でも完全活性化型構造の存在比が低かったことから、細胞外側ポケットを標的とする化合物では、MORの細胞内側に空間が開いた構造を安定化できないことが示唆されました。一方で、アロステリック薬剤であるBMS-986122がさらに結合した状態では、完全活性化型構造の存在比が増加したことから、既存薬とは異なる部位を作用点とすることにより、細胞内側空間が開いた構造が安定化されることが示されました。この機構により、BMS-986122が結合した状態では既存薬の薬効度の上限を超えてMORが活性化することが示されました。
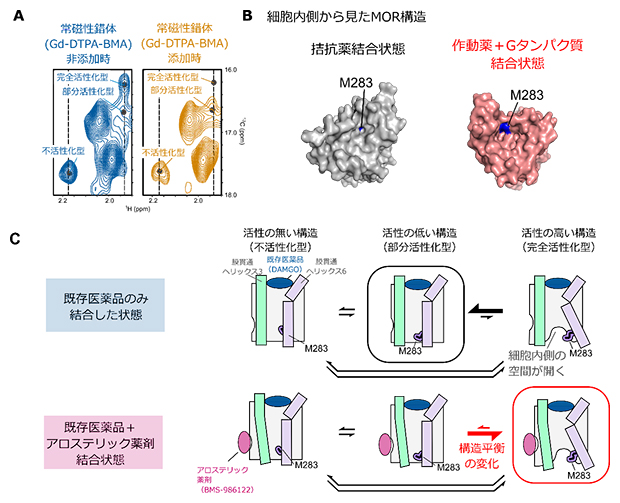
図3 アロステリック薬剤による構造平衡の変化
- A.完全作動薬(DAMGO)結合状態のMORにおける常時性錯体Gd-DTPA-BMA添加前後におけるM283のNMRシグナルの比較。Gd-DTPA-BMAを添加すると、完全活性化型のシグナルが常磁性緩和促進効果により減弱した。
- B.M283のMOR構造中における位置。M283の分子表面を青色で示した。M283は細胞内側に空間が開いた構造(赤)では、分子表面が露出する位置に存在する。
- C.アロステリック薬剤(BMS-986122)によるMORの活性化機構のモデル図。BMS-986122が結合すると構造平衡が変化し、細胞内側に空間が開いた構造の完全活性化型の存在比が増加する。
今後の期待
本研究により、細胞外側のポケットを標的とした既存薬では、細胞内側に空間が開いた構造を安定化できないために、MORの活性を十分に引き出せていないこと、および既存薬とは異なる部位に結合するアロステリック薬剤が結合すると、細胞内側に空間が開いた構造を安定化して既存薬の限界を超えてMORの活性が引き出されることが明らかになりました。この機構が明らかになったことにより、アロステリック薬剤をベースにした新規医薬品の合理的な開発が加速するものと期待できます。
多くのGPCRにおいて、その細胞外側のポケットを標的とした医薬品は、GPCRの動的な構造平衡を完全に活性化型に偏らせることができないことが分かっていましたが、本研究成果により、アロステリック薬剤がこうしたGPCRに対してその構造平衡状態を変え、既存薬の限界を超えて活性を上昇させることができる可能性が示されました。このことから、本研究の成果は、これまで十分な薬効を持つ医薬品が見つかっていないGPCR関連疾病に対し、十分な薬効を示す新しい医薬品開発に貢献することが期待できます。
補足説明
- 1.Gタンパク質共役型受容体(GPCR)、Gタンパク質
細胞膜表面に発現し、細胞外からの刺激を受容して、細胞内のGタンパク質を活性化することで細胞応答を引き起こす膜タンパク質の総称。細胞膜をαヘリックスが7回貫通する構造を特徴とする。GPCRはG protein-coupled receptorの略。Gタンパク質は、グアニンヌクレオチド結合タンパク質の総称。グアノシン二リン酸(GDP)が結合している不活性型と、グアノシン三リン酸(GTP)が結合している活性型の間でタンパク質のコンフォメーションが変化し、分子スイッチとして働く。 - 2.アロステリックモジュレーター(アロステリック薬剤)
受容体上において、生理的リガンドや医薬品が結合する部位から離れた領域に結合することで、その受容体のシグナル伝達活性を制御する化合物の総称。受容体に対して生理的リガンドや医薬品と同時に結合することで、協奏的に受容体に作用することを特徴とする。 - 3.溶液核磁気共鳴(NMR)法
溶液中の生体分子などを、強い磁場中に置くことで生じる原子核の共鳴現象を観測することで、生体分子の構造や運動性を原子レベルで調べることができる分光法。NMRはNuclear Magnetic Resonanceの略。 - 4.μオピオイド受容体(MOR)
鎮痛作用に関連した代表的なGPCR。モルヒネなどのオピオイド鎮痛薬が結合することにより活性化して、細胞内側へシグナルを伝達し鎮痛作用をもたらす。MORはmu opioid receptorの略。 - 5.常磁性錯体、常磁性緩和促進効果
孤立電子の電子スピンなどが持つ常磁性と呼ばれる性質を示す金属イオンを配位した錯体を常磁性錯体と呼ぶ。常磁性緩和促進効果は、常磁性錯体によって、核スピンの緩和速度が増大する効果のこと。ここでは、常磁性錯体であるGd-DTPA-BMAを溶媒に添加することによって、タンパク質中のNMRシグナルが、溶媒に分布した常磁性錯体からの距離の6乗に反比例して広幅化することを指す。
共同研究チーム
理化学研究所 生命機能科学研究センター 生体分子動的構造研究チーム
チームリーダー 嶋田 一夫(しまだ いちお)
(次世代天然物化学技術研究組合 技術顧問)
上級研究員 今井 駿輔(いまい しゅんすけ)
研修生 金子 舜(かねこ しゅん)
研修生 浅尾 信央(あさお のぶあき)
東京大学 大学院薬学系研究科 生命物理化学教室
准教授 上田 卓見(うえだ たくみ)
助教 幸福 裕(こうふく ゆたか)
研究支援
本研究は、理化学研究所運営費交付金(生命機能科学研究)で実施し、日本医療研究開発機構(AMED)「次世代治療・診断実現のための創薬基盤技術開発事業」(革新的中分子創薬技術の開発)および日本学術振興会(JSPS)科学研究費補助金特別推進研究「核磁気共鳴法による膜タンパク質のin situ機能解明(研究代表者:嶋田一夫)」による支援を受けて行われました。
原論文情報
- Shun Kaneko, Shunsuke Imai, Nobuaki Asao, Yutaka Kofuku, Takumi Ueda, and Ichio Shimada, "Activation mechanism of the μ-opioid receptor by an allosteric modulator", Proceedings of the National Academy of Sciences of the United States of America(PNAS), 10.1073/pnas.2121918119
発表者
理化学研究所
生命機能科学研究センター 生体分子動的構造研究チーム
チームリーダー 嶋田 一夫(しまだ いちお)
(次世代天然物化学技術研究組合 技術顧問)
上級研究員 今井 駿輔(いまい しゅんすけ)
研修生 金子 舜(かねこ しゅん)
 今井 駿輔
今井 駿輔
 金子 舜
金子 舜
報道担当
理化学研究所 広報室 報道担当
お問い合わせフォーム
AMED事業に関する問い合わせ
日本医療研究開発機構
創薬事業部 医薬品研究開発課
次世代治療・診断実現のための創薬基盤技術開発事業担当
Email: jisedai-med [at] amed.go.jp
次世代天然物化学技術研究組合に関する問い合わせ
次世代天然物化学技術研究組合問い合わせ窓口
Email: natprodchem [at] jbic.or.jp
※上記の[at]は@に置き換えてください。